Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Bindu Diana Paul | + 2332 word(s) | 2332 | 2021-09-23 06:16:26 | | | |
| 2 | Vivi Li | Meta information modification | 2332 | 2021-10-08 05:38:55 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Paul, B. Golgi apparatus and Huntington’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/14805 (accessed on 23 July 2026).
Paul B. Golgi apparatus and Huntington’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/14805. Accessed July 23, 2026.
Paul, Bindu. "Golgi apparatus and Huntington’s Disease" Encyclopedia, https://encyclopedia.pub/entry/14805 (accessed July 23, 2026).
Paul, B. (2021, October 01). Golgi apparatus and Huntington’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/14805
Paul, Bindu. "Golgi apparatus and Huntington’s Disease." Encyclopedia. Web. 01 October, 2021.
Copy Citation
Huntington’s disease (HD) is caused by expansion of polyglutamine repeats in the protein huntingtin, which affects the corpus striatum of the brain. The polyglutamine repeats in mutant huntingtin cause its aggregation and elicit toxicity by affecting several cellular processes, which include dysregulated organellar stress responses. The Golgi apparatus not only plays key roles in the transport, processing, and targeting of proteins, but also functions as a sensor of stress, signaling through the Golgi stress response. Unlike the endoplasmic reticulum (ER) stress response, the Golgi stress response is relatively unexplored. We have identified a Golgi stress response pathway which is dysregulated in HD.
Golgi apparatus
Huntington’s disease
cysteine
transsulfuration
Golgi stress response
integrated stress response
1. Introduction
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease which profoundly affects the corpus striatum of the brain; it results from expansion of polyglutamine repeats in the protein huntingtin [1]. Mutant huntingtin (mHtt) aggregates and affects cellular processes in multiple ways [2]. mHtt affects basic neuronal processes such as transcription, translation, nuclear-cytoplasmic transport, redox homeostasis, mitochondrial function and amino acid metabolism in addition to a myriad of physiological processes [3][4][5][6][7].
HD has also been linked to impaired stress responses involving redox homeostasis and endoplasmic stress response [5][7]. In addition to essential functions in cellular function, organelles serve important roles as sensors of stress and as hubs for signaling pathways. For instance, the endoplasmic reticulum (ER) plays central roles in protein folding, post-translational modifications, quality control of proteins and Ca2+ handling, among many other functions [8][9][10][11]. During ER stress—a state of functional imbalance—adaptive and restorative programs such as the unfolded protein response (UPR) and ER-associated protein degradation (ERAD), or autophagy, come into play [12][13]. One stimulus that triggers the ER stress response is the accumulation of unfolded or misfolded proteins in the ER lumen. Three arms exist in the ER stress response: the protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1) pathways, where each of the sensor proteins is a membrane protein (Figure 1). In the PERK arm, during stress, PERK dissociates from the chaperone protein, binding immunoglobulin protein/glucose-regulated protein 78 (BiP/GRP78), and undergoes dimerization and phosphorylation. PERK, (a component of the integrated stress response) then phosphorylates the eukaryotic translation initiation factor 2 subunit −α (eIF2α), which results in global translational arrest. Under these conditions only certain mRNAs such as those encoding activating transcription factor 4 (ATF4) are translated, in order to maintain functions important for cell survival. ATF4 regulates amino acid homeostasis, purine metabolism, response to oxidative stress, autophagy and apoptosis.
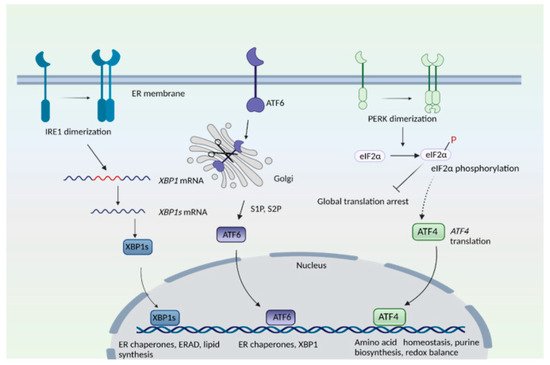
Figure 1. The endoplasmic reticulum (ER) stress response. The mammalian ER stress response consists of three arms: the inositol-requiring enzyme 1 (IRE1), protein kinase R (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6) pathways. IRE1 senses ER stress, which leads to its dimerization and to the activation of its endonuclease role, that is, to splice a specific intron from the mRNA of X-box binding protein 1, XBP1 to create XBP1s. T XBP1s protein translocates to the nucleus and transactivates its target genes. In the ATF6 arm, binding immunoglobulin protein/glucose-regulated protein 78 (BiP/GRP78) dissociates from ATF6 when unfolded proteins accumulate in the ER. ATF6 translocates to the Golgi complex, where it undergoes proteolytic cleavage by site 1 and site 2 proteases (S1P and S2P). The N-terminal cytosolic fragment of ATF6 migrates to the nucleus and induces expression of target genes. In the PERK arm, dissociation of BiP causes its dimerization and autophosphorylation. PERK then phosphorylates eukaryotic initiation factor 2α (eIF2α), which results in global translational arrest. Under these conditions, only certain mRNAs such as ATF4 are translated, in order to maintain cellular functions during stress.
In the inositol-requiring enzyme (IRE) branch, the ER-resident IRE1 senses unfolded proteins or lipid disequilibrium and undergoes dimerization and autophosphorylation, activating IRE1′s cytosolic endonuclease domain, which then splices a specific intron from the mRNA of X-box binding protein 1u, XBP1u to create XBP1s. The XBP1s protein translocates to the nucleus and transactivates genes involved in protein degradation, protein folding, and lipid metabolism [14][15]. The third arm of the UPR consists of ATF6, an ER transmembrane protein that translocates to the Golgi when activated. During ER stress, when unfolded proteins accumulate, BiP/GRP78 dissociates from ATF6 to cause translocation of ATF6 into the Golgi. In the Golgi, site 1 protease (S1P) and site 2 protease (S2P) cleave ATF6 [16]. The N-terminal region of ATF6 functions as a transcription factor and stimulates expression of target genes, such as protein disulfide isomerase (PDI), XBP1, and C/EBP Homologous Protein (CHOP) [16][17]. When proteins cannot be repaired or folded back into their functional configurations, they are targeted for degradation by ERAD [18]. When the repair capacity of ERAD is crossed, portions of the ER can be specifically targeted for degradation through autophagy (ER-phagy) [19]. Recently, we elucidated the involvement of signaling pathways modulated by the Golgi apparatus in HD [20][21].
2. Golgi Stress Response and Redox Imbalance in Neurodegeneration: Focus on Huntington’s Disease
Accumulating evidence reveals that abnormalities in the structure and function of Golgi apparatus occur in neurodegenerative diseases including Alzheimer’s disease (AD), Amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), Huntington’s disease (HD) and Creutzfeldt–Jakob disease [20][22][23][24][25][26][27]. The Golgi has also been reported to be fragmented during viral infection [28]. Depletion of the golgin GM130 has been reported to cause Golgi disruption, Purkinje neuron loss, and ataxia in mice [29]. Golgi fragmentation in dopaminergic neurons in the substantia nigra has been observed in Parkinson’s disease patients [30]. Early studies revealed that the Golgi apparatus may be fragmented in a population of neurons without neurofibrillary tangles (NFTs) [31]. In JNPL3 transgenic mice—which express the P301L mutant of tau, a component of NFTs and paired helical filaments (PHFs)—the Golgi complex was fragmented; however, mitochondria or other membranous organelles appeared normal, indicating that Golgi fragmentation is one of the earliest events that occur during pathogenesis, a finding which has been suggested by other laboratories as well [32][33][34]. Structural deformities in the Golgi complex were also linked to accumulation of phospho-tau in the P301S mouse model of AD [35]. Aging is a major risk factor for neurodegeneration; not surprisingly, increased Golgi fragmentation in neurons was observed with aging. GRASP65 and Golgin-84 were also diminished in the aging mouse brain [36].
In HD, we showed that elevated levels of ACBD3 occurred in cell culture and mouse models as well as human HD [20]. ACBD3 bound to the striatal protein ras homolog enriched in striatum (Rhes), which binds to mutant huntingtin (mHtt) and mediates cell death in HD [37]. More recently, we identified another arm of the Golgi stress signaling pathway in a striatal progenitor cell line model of HD. HD is a neurodegenerative disorder triggered by expansion of CAG repeats (encoding polyglutamine repeats) in the gene encoding huntingtin and which primarily affects the corpus striatum of the brain, manifesting as abnormal involuntary movements along with motor and cognitive deficits [1][2]. mHtt affects multiple cellular processes such as DNA replication and repair, transcription, translation, nucleocytoplasmic trafficking, mitochondrial function and proteostasis, to name a few [4][5][6][38][39][40][41][42].
2.1. Redox Imbalance and Cysteine Metabolism in HD
A hallmark of HD is increased oxidative stress. Oxidative stress occurs when the balance between prooxidant and antioxidant pathways tilts in favor of the former. However, oxidative stress has more recently been defined as a disruption of redox signaling pathways [43]. Elevated oxidative stress is at the heart of several neurodegenerative diseases, as well as other conditions [44][45]. Decreased levels and/or dysregulated metabolism of the antioxidants such as ascorbate (vitamin C), glutathione (GSH) and cysteine, and coenzyme Q10 (CoQ10) have been observed in HD and contribute to disease pathology [46][47][48]. Both biosynthesis and the uptake of cysteine or its oxidized form cystine are compromised in HD, causing elevated oxidative stress [5][49][50]. The activity of the neuronal cysteine transporter EAAT3/EAAC1 is reduced in HD due to decreased trafficking to the cell membrane [51]. Decreased expression of cystathionine γ-lyase (CSE) (the biosynthetic enzyme for cysteine) also occurs in HD as mHtt sequesters specificity protein 1 (SP1), the transcription factor for CSE during basal conditions [47][52][53]. CSE is also regulated by activating transcription factor 4 (ATF4) in response to stress. In HD-affected cells the induction of ATF4 is suboptimal, leading to decreased CSE expression and cysteine biosynthesis [5]. Cysteine is utilized in the biosynthesis of sulfur-containing molecules such as coenzyme A, taurine, lanthionine, homolanthionine, and cystamine [54]. It is also the substrate for production of the gaseous signaling molecule hydrogen sulfide (H2S) [55][56]. H2S signals by sulfhydration or persulfidation, a posttranslational modification which occurs on the –SH group of reactive cysteine residues, leading to formation of –SSH or persulfide groups [57]. Sulfhydration modulates the function of several proteins and signaling cascades, including response to inflammation, mitochondrial bioenergetics and stress responses [58][59]. Both cysteine metabolism and sulfhydration are altered in HD, which contributes to increased protein oxidation [5][47][60].
2.2. Golgi Stress Response and Links to Redox Homeostasis
ATF4, a master regulator of amino acid homeostasis and stress responses, is a central player in the integrated stress response [61][62][63]. It also regulates purine biosynthesis and regulates mTOR function [64]. Furthermore, ATF4 modulates the switch from synthesis of fetal hemoglobin to adult hemoglobin by stimulating transcription of BCL11A, a repressor of γ-globin synthesis [65]. ATF4 harbors a basic leucine-zipper (bZIP) domain and can either form homodimers or heterodimerize with other members of the bZIP family (FOS/JUN, ATF and CCAAT enhancer-binding protein (C/EBP) bZIP transcription factors) to control transcription [66][67]. Nuclear factor erythroid 2-related factor 2 (Nrf2), the master regulator of redox regulation, also forms heterodimers with ATF4 and stimulates the transcription of cytoprotective genes during oxidative stress [68]. ATF4 may have dual functions, modulating either cell survival or cell death, and excessive stimulation of ATF4 signaling may cause cell death [69]. The pro-survival or apoptotic function of ATF4 has been attributed in part to the identity of its heterodimerization partner and the physiologic context; the mechanisms are still being elucidated [70].
Expression of ATF4 is regulated at the transcriptional as well as the translational level, and is a vital part of the integrated stress response [71][72]. The integrated stress response (Figure 2) is engaged in response to stress stimuli, including but not limited to amino acid and nutrient deprivation, ER stress, mitochondrial stress, iron dysregulation and viral infection. Four kinases, namely general control non-derepressible 2 (GCN2), PKR-like ER kinase (PERK) 19, double-stranded RNA-dependent protein kinase (PKR), and heme-regulated eIF2α kinase (HRI) sense the stress and phosphorylate the eukaryotic initiation 2 α (eIF2α), which abrogates its catalytic activity resulting in global translational arrest [61][73][74][75][76][77]. Under these conditions, only mRNAs responsible for maintenance of cell survival and essential signaling (such as ATF4) are translated. Recently, we showed that the Golgi stressors, monensin and nigericin, activated the integrated stress response by eliciting phosphorylation of PERK, resulting in translation of ATF4 and expression of its targets [21] (Figure 3). Among these targets were enzymes involved in cysteine biosynthesis and uptake, cystathionine γ-lyase (CSE) and SLC7A11, a subunit of the XcT transporter, which imports cystine, the oxidized form of cysteine (Figure 3a).
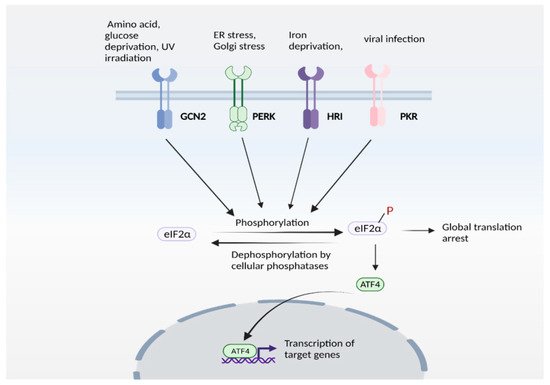
Figure 2. The integrated stress response (ISR). ISR is activated by several stress stimuli. In mammals, these are sensed by four kinases: general control non-depressible 2 (GCN2), protein kinase R protein kinase R (PKR)-like ER kinase (PERK), Heme-regulated eIF2α kinase (HRI) and double stranded RNA dependent protein kinase (PKR), each of which respond to a set of stimuli. These kinases undergo autophosphorylation and phosphorylate the eukaryotic initiation factor 2α (eIF2α), which inhibits its catalytic activity and its function of delivering the initiator tRNA to the ribosome, thereby arresting global translation. Under these conditions, only certain mRNAs (such as that encoding ATF4) are translated. ATF4 functions either as a homodimer or heterodimer to transactivate its target genes. The pathway is referred to as ISR because signaling mediated by diverse stress stimuli converge upon a common pathway (eIF2α/ATF4 axis).
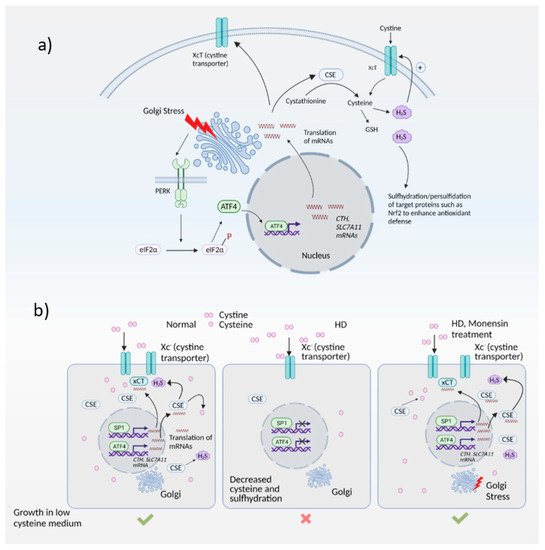
Figure 3. The Golgi stress response and its intersection with redox homeostasis in Huntington’s disease (HD). (a) Golgi stress response in normal cells. Golgi stress activates PERK, which phosphorylates eIF2α to inhibit general protein synthesis. Only mRNAs such as ATF4 are translated. ATF4 regulates amino acid homeostasis and one of the genes induced by ATF4 is CTH (which encodes the biosynthetic enzyme for cysteine, also called CSE). CSE utilizes cysteine to produce the gaseous signaling molecule hydrogen sulfide (H2S). H2S signals by a post-translational modification termed sulfhydration/persulfidation and modulates the activity of target proteins. H2S stimulates cystine uptake by the cystine transporter, leading to increased cysteine levels in cells. ATF4 also regulates expression of SLC7A11 (xCT), a subunit of the cystine transporter, by activating its transcription through heterodimerization with Nrf2, a master regulator of redox homeostasis. (b) Harnessing the Golgi stress response to elicit cytoprotection in HD. Normal cells express CSE and ATF4 during basal conditions and during stress to produce cysteine. Cysteine is also imported into cells via the cystine transporter, Xc-. In HD, both basal expression of CSE (regulated by specificity protein1, SP1) as well as stress-induced expression of CSE and the xCT subunit of the cystine transporter by ATF4 are compromised, causing a cysteine deficit which leads to decreased H2S levels and sulfhydration. When cells are treated with monensin, a Golgi stressor, CSE is induced via the PERK/ATF4 pathway to increase cysteine and H2S levels and mediate cytoprotection.
In the striatal progenitor cell culture models STHdhQ7/Q7 (Q7) and STHdh1117/Q111 (Q111), treatment with monensin rescued cell death associated with cysteine deprivation. Monensin increased the expression of ATF4 in the Q111 cells, which have compromised stress responses. We had shown previously that Q111 cells had decreased expression of CSE and thus could not grow in the absence of cysteine [5][47]. These cells were also compromised in their ability to upregulate ATF4, the transcription factor responsible for induction of CSE during cysteine deprivation. Treatment with monensin rescued growth in cysteine-free media and decreased oxidative stress in a manner dependent on PERK (Figure 3b). Monensin failed to upregulate ATF4 and CSE expression in cells deleted for PERK. Thus, Golgi stress engages the PERK pathway in HD cells. We further showed that mild Golgi stress can be harnessed to elicit cytoprotective effects [21].
References
- O’Donovan, M.C. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983.
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005.
- Ahmed, I.; Sbodio, J.I.; Harraz, M.M.; Tyagi, R.; Grima, J.C.; Albacarys, L.K.; Hubbi, M.; Xu, R.; Kim, S.; Paul, B.D.; et al. Huntington’s disease: Neural dysfunction linked to inositol polyphosphate multikinase. Proc. Natl. Acad. Sci. USA 2015, 112, 9751–9756.
- Grima, J.C.; Daigle, J.G.; Arbez, N.; Cunningham, K.; Zhang, K.; Ochaba, J.; Geater, C.; Morozko, E.; Stocksdale, J.; Glatzer, J.C.; et al. Mutant Huntingtin Disrupts the Nuclear Pore Complex. Neuron 2017, 94, 93–107.
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Transcriptional control of amino acid homeostasis is disrupted in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2016, 113, 8843–8848.
- Eshraghi, M.; Karunadharma, P.P.; Blin, J.; Shahani, N.; Ricci, E.P.; Michel, A.; Urban, N.T.; Galli, N.; Sharma, M.; Ramírez-Jarquín, U.N.; et al. Mutant Huntingtin stalls ribosomes and represses protein synthesis in a cellular model of Huntington disease. Nat. Commun. 2021, 12, 1–20.
- Paul, B.D.; Snyder, S.H. Impaired Redox Signaling in Huntington’s Disease: Therapeutic Implications. Front. Mol. Neurosci. 2019, 12, 68.
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox. Signal 2014, 21, 396–413.
- Reid, D.W.; Nicchitta, C.V. Diversity and selectivity in mRNA translation on the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2015, 16, 221–231.
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell Mol. Life Sci. 2016, 73, 79–94.
- Roscoe, J.M.; Sevier, C.S. Pathways for Sensing and Responding to Hydrogen Peroxide at the Endoplasmic Reticulum. Cells 2020, 9, 2314.
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 2021, 47, 101169.
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438.
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086.
- Frakes, A.E.; Dillin, A. The UPR(ER): Sensor and Coordinator of Organismal Homeostasis. Mol. Cell 2017, 66, 761–771.
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER Stress Induces Cleavage of Membrane-Bound ATF6 by the Same Proteases that Process SREBPs. Mol. Cell 2000, 6, 1355–1364.
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian Transcription Factor ATF6 Is Synthesized as a Transmembrane Protein and Activated by Proteolysis in Response to Endoplasmic Reticulum Stress. Mol. Biol. Cell 1999, 10, 3787–3799.
- Lemberg, M.K.; Strisovsky, K. Maintenance of organellar protein homeostasis by ER-associated degradation and related mechanisms. Mol. Cell 2021, 81, 2507–2519.
- Wilkinson, S. Emerging Principles of Selective ER Autophagy. J. Mol. Biol. 2020, 432, 185–205.
- Sbodio, J.I.; Paul, B.D.; Machamer, C.E.; Snyder, S.H. Golgi Protein ACBD3 Mediates Neurotoxicity Associated with Huntington’s Disease. Cell Rep. 2013, 4, 890–897.
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Golgi stress response reprograms cysteine metabolism to confer cytoprotection in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 780–785.
- Gonatas, N.K.; Stieber, A.; Gonatas, J.O. Fragmentation of the Golgi apparatus in neurodegenerative diseases and cell death. J. Neurol. Sci. 2006, 246, 21–30.
- Mourelatos, Z.; Gonatas, N.K.; Stieber, A.; Gurney, M.E.; Canto, M.C.D. The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu, Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc. Natl. Acad. Sci. USA 1996, 93, 5472–5477.
- Joshi, G.; Bekier, M.E., 2nd; Wang, Y. Golgi fragmentation in Alzheimer’s disease. Front. Neurosci. 2015, 9, 340.
- Rendón, W.O.; Martínez-Alonso, E.; Tomás, M.; Martínez-Martínez, N.; Martínez-Menárguez, J.A. Golgi fragmentation is Rab and SNARE dependent in cellular models of Parkinson’s disease. Histochem. Cell Biol. 2012, 139, 671–684.
- Strehlow, A.N.; Li, J.; Myers, R.M. Wild-type huntingtin participates in protein trafficking between the Golgi and the extracellular space. Hum. Mol. Genet. 2006, 16, 391–409.
- Sakurai, A.; Okamoto, K.; Fujita, Y.; Nakazato, Y.; Wakabayashi, K.; Takahashi, H.; Gonatas, N.K. Fragmentation of the Golgi apparatus of the ballooned neurons in patients with corticobasal degeneration and Creutzfeldt-Jakob disease. Acta Neuropathol. 2000, 100, 270–274.
- Campadelli, G.; Brandimarti, R.; Di Lazzaro, C.; Ward, P.L.; Roizman, B.; Torrisi, M.R. Fragmentation and dispersal of Golgi proteins and redistribution of glycoproteins and glycolipids processed through the Golgi apparatus after infection with herpes simplex virus. Proc. Natl. Acad. Sci. USA 1993, 90, 2798–2802.
- Liu, C.; Mei, M.; Li, Q.; Roboti, P.; Pang, Q.; Ying, Z.; Gao, F.; Lowe, M.; Bao, S. Loss of the golgin GM130 causes Golgi disruption, Purkinje neuron loss, and ataxia in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 346–351.
- Tomas, M.; Martinez-Alonso, E.; Martinez-Martinez, N.; Cara-Esteban, M.; Martinez-Menarguez, J.A. Fragmentation of the Golgi complex of dopaminergic neurons in human substantia nigra: New cytopathological findings in Parkinson’s disease. Histol. Histopathol. 2021, 36, 47–60.
- Stieber, A.; Mourelatos, Z.; Gonatas, N.K. In Alzheimer’s disease the Golgi apparatus of a population of neurons without neurofibrillary tangles is fragmented and atrophic. Am. J. Pathol. 1996, 148, 415–426.
- Liazoghli, D.; Perreault, S.; Micheva, K.D.; Desjardins, M.; Leclerc, N. Fragmentation of the Golgi Apparatus Induced by the Overexpression of Wild-Type and Mutant Human Tau Forms in Neurons. Am. J. Pathol. 2005, 166, 1499–1514.
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn, K.; Murphy, M.P.; Baker, M.; Yu, X.; et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000, 25, 402–405.
- Nakagomi, S.; Barsoum, M.J.; Bossy-Wetzel, E.; Sütterlin, C.; Malhotra, V.; Lipton, S.A. A Golgi fragmentation pathway in neurodegeneration. Neurobiol. Dis. 2008, 29, 221–231.
- Antón-Fernández, A.; Merchán-Rubira, J.; Avila, J.; Hernández, F.; DeFelipe, J.; Muñoz, A. Phospho-Tau Accumulation and Structural Alterations of the Golgi Apparatus of Cortical Pyramidal Neurons in the P301S Tauopathy Mouse Model. J. Alzheimer’s Dis. 2017, 60, 651–661.
- Jiang, Q.; Wang, L.; Guan, Y.; Xu, H.; Niu, Y.; Han, L.; Wei, Y.-P.; Lin, L.; Chu, J.; Wang, Q.; et al. Golgin-84-associated Golgi fragmentation triggers tau hyperphosphorylation by activation of cyclin-dependent kinase-5 and extracellular signal-regulated kinase. Neurobiol. Aging 2014, 35, 1352–1363.
- Subramaniam, S.; Sixt, K.M.; Barrow, R.; Snyder, S.H. Rhes, a Striatal Specific Protein, Mediates Mutant-Huntingtin Cytotoxicity. Science 2009, 324, 1327–1330.
- Lu, X.H.; Mattis, V.B.; Wang, N.; Al-Ramahi, I.; van den Berg, N.; Fratantoni, S.A.; Waldvogel, H.; Greiner, E.; Osmand, A.; Elzein, K.; et al. Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci. Transl. Med. 2014, 6, 268ra178.
- Iyer, R.R.; Pluciennik, A. DNA Mismatch Repair and its Role in Huntington’s Disease. J. Huntingtons Dis. 2021, 10, 75–94.
- Luthi-Carter, R.; Hanson, S.A.; Strand, A.D.; Bergstrom, D.A.; Chun, W.; Peters, N.L.; Woods, A.M.; Chan, E.Y.; Kooperberg, C.; Krainc, D.; et al. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: Parallel changes in muscle and brain. Hum. Mol. Genet. 2002, 11, 1911–1926.
- Bañez-Coronel, M.; Ayhan, F.; Tarabochia, A.D.; Zu, T.; Perez, B.A.; Tusi, S.K.; Pletnikova, O.; Borchelt, D.R.; Ross, C.A.; Margolis, R.L.; et al. RAN Translation in Huntington Disease. Neuron 2015, 88, 667–677.
- Soares, T.R.; Reis, S.D.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M.A. Targeting the proteostasis network in Huntington’s disease. Ageing Res. Rev. 2019, 49, 92–103.
- Jones, D.P. Redefining Oxidative Stress. Antioxid. Redox Signal. 2006, 8, 1865–1879.
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Op-portunities. Antioxid. Redox Signal. 2019, 30, 1450–1499.
- Paul, B.D.; Lemle, M.D.; Komaroff, A.L.; Snyder, S.H. Redox imbalance links COVID-19 and myalgic encephalomyelitis/chronic fatigue syndrome. Proc. Natl. Acad. Sci. USA 2021, 118, e2024358118.
- Andrich, J.; Saft, C.; Gerlach, M.; Schneider, B.; Arz, A.; Kühn, W.; Müller, T. Coenzyme Q10 serum levels in Huntington’s disease. In Focus on Extrapyramidal Dysfunction; Springer: Vienna, Austria, 2004; Volume 68, pp. 111–116.
- Paul, B.D.; Sbodio, J.I.; Xu, R.; Vandiver, M.S.; Cha, J.Y.; Snowman, A.M.; Snyder, S.H. Cystathionine gamma-lyase deficiency mediates neurodegeneration in Huntington’s disease. Nature 2014, 509, 96–100.
- Acuña, A.I.; Esparza, M.; Kramm, C.; Beltrán, F.A.; Parra, A.V.; Cepeda, C.; Toro, C.A.; Vidal, R.L.; Hetz, C.; Concha, I.I.; et al. A failure in energy metabolism and antioxidant uptake precede symptoms of Hun-tington’s disease in mice. Nat. Commun. 2013, 4, 2917.
- Zhang, C.; Rodriguez, C.; Spaulding, J.; Aw, T.Y.; Feng, J. Age-dependent and tissue-related glutathione redox status in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2012, 28, 655–666.
- Frederick, N.M.; Bertho, J.; Patel, K.K.; Petr, G.T.; Bakradze, E.; Smith, S.B.; Rosenberg, P.A. Dysregulation of system xc(-) expression induced by mutant huntingtin in a striatal neuronal cell line and in R6/2 mice. Neurochem. Int. 2014, 76, 59–69.
- Li, X.; Valencia, A.; Sapp, E.; Masso, N.; Alexander, J.; Reeves, P.; Kegel, K.B.; Aronin, N.; DiFiglia, M. Aberrant Rab11-dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington’s disease. J. Neurosci. 2010, 30, 4552–4561.
- Dunah, A.W.; Jeong, H.; Griffin, A.; Kim, Y.M.; Standaert, D.G.; Hersch, S.M.; Mouradian, M.M.; Young, A.B.; Tanese, N.; Krainc, D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science 2002, 296, 2238–2243.
- Paul, B.D.; Snyder, S.H. Neurodegeneration in Huntington’s disease involves loss of cystathionine gamma-lyase. Cell Cycle 2014, 13, 2491–2493.
- Paul, B.D.; Sbodio, J.I.; Snyder, S.H. Cysteine Metabolism in Neuronal Redox Homeostasis. Trends Pharmacol. Sci. 2018, 39, 513–524.
- Paul, B.D.; Snyder, S.H. H2S: A Novel Gasotransmitter that Signals by Sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700.
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions. Mol. Cell 2012, 45, 13–24.
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S Signals Through Protein S-Sulfhydration. Sci. Signal. 2009, 2, ra72.
- Paul, B.D.; Snyder, S.H. Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem. Pharmacol. 2018, 149, 101–109.
- Paul, B.D.; Snyder, S.H.; Kashfi, K. Effects of hydrogen sulfide on mitochondrial function and cellular bioenergetics. Redox Biol. 2021, 38, 101772.
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, J.L.; Thomas-Lopez, D.; Jung, Y.; et al. Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metab. 2019, 30, 1152–1170.e13.
- Harding, H.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.S.; et al. An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Mol. Cell 2003, 11, 619–633.
- Kilberg, M.S.; Shan, J.; Su, N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 2009, 20, 436–443.
- Rzymski, T.; Milani, M.; Pike, L.; Buffa, F.; Mellor, H.R.; Winchester, L.; Pires, I.; Hammond, E.; Ragoussis, I.; Harris, A.L. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010, 29, 4424–4435.
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733.
- Huang, P.; Peslak, S.A.; Lan, X.; Khandros, E.; Yano, J.A.; Sharma, M.; Keller, C.A.; Giardine, B.M.; Qin, K.; Abdulmalik, O.; et al. HRI-regulated transcription factor ATF4 activates BCL11A transcription to silence fetal hemoglobin expression. Blood 2020, 135, 2121–2132.
- Huggins, C.J.; Mayekar, M.K.; Martin, N.; Saylor, K.L.; Gonit, M.; Jailwala, P.; Kasoji, M.; Haines, D.C.; Quiñones, O.A.; Johnson, P.F. C/EBPgamma Is a Critical Regulator of Cellular Stress Response Networks through Heterodimerization with ATF. Mol. Cell Biol. 2015, 36, 693–713.
- Hai, T.; Curran, T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc. Natl. Acad. Sci. USA 1991, 88, 3720–3724.
- He, C.H.; Gong, P.; Hu, B.; Stewart, D.; Choi, M.E.; Choi, A.M.K.; Alam, J. Identification of Activating Transcription Factor 4 (ATF4) as an Nrf2-interacting Protein. J. Biol. Chem. 2001, 276, 20858–20865.
- Lange, P.S.; Chavez, J.C.; Pinto, J.T.; Coppola, G.; Sun, C.W.; Townes, T.M.; Geschwind, D.H.; Ratan, R.R. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J. Exp. Med. 2008, 205, 1227–1242.
- Wortel, I.M.N.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving Stress: Modulation of ATF4-Mediated Stress Re-sponses in Normal and Malignant Cells. Trends Endocrinol. Metab. 2017, 28, 794–806.
- Dey, S.; Baird, T.; Zhou, D.; Palam, L.R.; Spandau, D.F.; Wek, R.C. Both Transcriptional Regulation and Translational Control of ATF4 Are Central to the Integrated Stress Response. J. Biol. Chem. 2010, 285, 33165–33174.
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395.
- Berlanga, J.J.; Herrero, S.; de Haro, C. Characterization of the hemin-sensitive eukaryotic initiation factor 2alpha kinase from mouse nonerythroid cells. J. Biol. Chem. 1998, 273, 32340–32346.
- Chen, J.J.; Throop, M.S.; Gehrke, L.; Kuo, I.; Pal, J.; Brodsky, M.; London, I.M. Cloning of the cDNA of the heme-regulated eukaryotic initiation factor 2 alpha (eIF-2 alpha) kinase of rabbit reticulocytes: Homology to yeast GCN2 protein kinase and human double-stranded-RNA-dependent eIF-2 alpha kinase. Proc. Natl. Acad. Sci. USA 1991, 88, 7729–7733.
- Shi, Y.; Vattem, K.M.; Sood, R.; An, J.; Liang, J.; Stramm, L.; Wek, R.C. Identification and Characterization of Pancreatic Eukaryotic Initiation Factor 2 α-Subunit Kinase, PEK, Involved in Translational Control. Mol. Cell. Biol. 1998, 18, 7499–7509.
- Meurs, E.; Chong, K.; Galabru, J.; Thomas, N.B.; Kerr, I.M.; Williams, B.; Hovanessian, A.G. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 1990, 62, 379–390.
- Donnelly, N.; Gorman, A.; Gupta, S.; Samali, A. The eIF2α kinases: Their structures and functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511.
More
Information
Subjects:
Medicine, Research & Experimental
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
3.8K
Revisions:
2 times
(View History)
Update Date:
08 Oct 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No