Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Daniel Sausen | + 4310 word(s) | 4310 | 2021-09-22 06:10:48 | | | |
| 2 | Bruce Ren | -13 word(s) | 4297 | 2021-09-30 04:04:47 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sausen, D. Stress-Induced Epstein-Barr Virus Reactivation. Encyclopedia. Available online: https://encyclopedia.pub/entry/14764 (accessed on 25 June 2026).
Sausen D. Stress-Induced Epstein-Barr Virus Reactivation. Encyclopedia. Available at: https://encyclopedia.pub/entry/14764. Accessed June 25, 2026.
Sausen, Daniel. "Stress-Induced Epstein-Barr Virus Reactivation" Encyclopedia, https://encyclopedia.pub/entry/14764 (accessed June 25, 2026).
Sausen, D. (2021, September 29). Stress-Induced Epstein-Barr Virus Reactivation. In Encyclopedia. https://encyclopedia.pub/entry/14764
Sausen, Daniel. "Stress-Induced Epstein-Barr Virus Reactivation." Encyclopedia. Web. 29 September, 2021.
Copy Citation
Epstein-Barr virus (EBV) is typically found in a latent, asymptomatic state in immunocompetent individuals. Perturbations of the host immune system can stimulate viral reactivation. Furthermore, there are a myriad of EBV-associated illnesses including various cancers, post-transplant lymphoproliferative disease, and autoimmune conditions. A thorough understanding of this virus, and the interplay between stress and the immune system, is essential to establish effective treatment.
EBV
latency
reactivation
stress
vaccination
1. Introduction
Epstein-Barr virus (EBV), also known as human herpesvirus 4, is a widely prevalent pathogen that infects 90% or more of the world population [1][2][3]. Infection most commonly occurs following exposure to contaminated oral secretions, although symptoms do not appear for approximately six weeks, if at all [1]. EBV establishes lifelong latency in infected lymphocytes following acute infection. It can reactivate under appropriate conditions, namely those associated with diminished cell-mediated immunity [4].
The primary disease associated with EBV is infectious mononucleosis. This illness is most commonly seen in adolescents and young adults and presents with fatigue, fever, pharyngitis, cervical lymphadenopathy, and lymphocytosis [5][6]. EBV has been associated with other diseases including chronic fatigue syndrome, Epstein-Barr virus-related post-transplant lymphoproliferative disease, multiple sclerosis, encephalitis, cerebellar ataxia, Alzheimer’s disease, oral hairy leukoplakia, and autoimmune conditions such as Grave’s Disease, Sjögren’s syndrome, and rheumatoid arthritis [6][7][8][9][10][11][12][13][14]. Notably, there is a well-established connection between EBV and malignancies including Hodgkin’s lymphoma, diffuse large B-cell lymphoma, Burkitt lymphoma, primary central nervous system lymphoma, T cell lymphoma, certain gastric carcinomas, and nasopharyngeal carcinoma (NPC) [15][16][17][18]. In fact, it is estimated that EBV causes 1.8% of all cancer-related deaths worldwide [19], while a more recent study found that 265,000 cases of Burkitt lymphoma, Hodgkin’s lymphoma, NPC, and gastric carcinoma alone were caused by EBV in 2017 [20]. There is no vaccine for EBV and current anti-EBV agents are suboptimal due to low potency or high toxicity [21][22].
EBV is a member of the gammaherpesvirus subfamily of herpesviridae. Its virion structure is similar to other herpesviridae and includes a double-stranded DNA core, a surrounding icosahedral capsid, a tegument, and an envelope studded with glycoproteins [23][24]. The tegument encompasses the area between the capsid and envelope [23]. The proteins found therein are involved in numerous viral processes, including reactivation [25], viral envelopment [26], and immune evasion [27][28]. Glycoproteins play an integral role in viral fusion and have also been implicated in immune system evasion [29].
EBV’s ability to infect B cells and epithelial cells is well established [30][31]. The core fusion machinery required for viral entry include the EBV glycoproteins (g) gB, gH, and gL [32]. In B cells, gH and gL complex with gp42 to form a gH/gL/gp42 heterotrimer that is necessary for entry [33]. gp42 is able to interact with human leukocyte antigen (HLA) class II molecules on B cells to trigger viral fusion [32]. gp220/350 tethers EBV to B cells via interactions with complement receptor type 2 (CD21) [34]. In epithelial cells, gH and gL form a heterodimer that can bind the epithelial cell integrins αVβ5, αVβ6, or αVβ8 in the early stages of viral entry [33]. BMRF2, another EBV glycoprotein, likewise interacts with cellular integrins, specifically α1, α5, α3, and αv integrins, to facilitate infection of polarized epithelial cells [35]. Other cellular factors that have been identified as important in EBV epithelial cell entry include neuropilin 1, which interacts with gB [36], ephrin receptor A2, which interacts with gH/gL and gB [37], and non-muscle myosin heavy chain IIA, which interacts with gH/gL [38].
The role of gp220/350 in epithelial cell infection is not as well established, and conflicting reports exist regarding its utility [39][40]. Notably, deleting gp220/350 did not completely abrogate EBV entry into numerous examined cell lines, including human B cells, lymphoid lines, and the majority of epithelial cell lines, although infection was not as efficient in the absence of gp220/350. This indicates that gp220/350 is not necessarily required for infection of either epithelial or lymphocyte cell lines [41]. EBV’s ability to infect T cells is less studied, though it was recently shown that CD21 is important in T cell entry [42]. EBV is also capable of infecting NK cells either by direct viral episome transfer [43] or by a CD21-dependent mechanism. In the latter case, NK cells targeting infected B cells temporarily gain CD21 molecules through synaptic transfer. This allows EBV to bind to and infect the NK cell [44]. A more thorough review of EBV tropism can be found elsewhere [45][46].
In vitro, EBV is capable of infecting numerous cell lines. Examples of B cell lines shown to sustain EBV infection include lymphoblastoid, P3HR1, Rael, Akata, Raji, Daudi, and B95-8 cells, while examples of epithelioid cell lines sustaining EBV infection include GT38, PN, the nasopharyngeal carcinoma line C666, and the gastric carcinoma line AGS [47][48][49][50][51]. EBV has also been shown to infect monocytes [52]. Indeed, an EBV-infected monocyte cell line called E1 has been established [53]. EBV’s ability to infect neuronal cells was established by Jha et al., who were able to successfully infect the neuroblastoma cell line Sh-Sy5y, neurons from the teratocarcinoma line Ntera2, and primary human fetal neurons [54]. In addition, EBV has been shown to infect HMC-3, a microglial cell line, and U-87 MG, an astrocyte cell line [55].
This review will begin with an overview of EBV reactivation and the lytic and latent cycles, including recent advances in our understanding of how EBV establishes and maintains latency. It will next explore how cellular and psychological stressors lead to EBV reactivation. We will conclude with a brief overview of advances in treatments targeting EBV.
2. Overview of EBV Reactivation and the Lytic Cycle
Herpesvirus lytic replication involves three stages of gene expression: immediate early, early, and late [56]. The transcription factors BZLF1, also known as Zta, ZEBRA, EB1, or Z, and BRLF1, also known as Rta or R, are critical to the reactivation of the lytic cycle. As the master regulator, BZLF1 is particularly important in this activation [57]. These two genes induce the other’s expression [58]; in fact, one key role of ZEBRA, the protein encoded by BZLF1, is to stimulate the BRLF1 gene, which leads to the production of the protein Rta [57]. Transcription from oriLyt requires BZLF1 and BRLF1 expression. Expression of the early gene BSMLF1, also known as SM, Mta, and EB2, is also essential in gene transcription. The protein encoded by this gene has been shown to both upregulate lytic gene synthesis and downregulate host protein synthesis by impacting mRNA stability and transport through a direct interaction with the RNA [59][60]. BMRF1 is another key early protein that interacts with the BALF 5 DNA polymerase subunit to enhance nucleotide processivity. It does this by stabilizing the interaction between the primer template and the polymerase [61]. Early genes have diverse functional roles outside of replication as well. For example, BARF1 is an early gene involved in immune modulation. Specifically, it was found to inhibit colony-stimulating factor-1 activity through mimicry of the colony-stimulating factor-1 receptor c-fms [62]. BHRF2 is another EBV early gene that exerts its effect through molecular mimicry. It was shown to resemble the human antiapoptotic protein Bcl-2. Like Bcl-2, it can improve B cell survival [63].
Requirements for late gene viral transcription include viral DNA replication [64][65] and the interaction between the EBV-encoded BCRF1 protein and a viral pentamer with cellular RNA polymerase II [66]. The late phase of the lytic cycle involves the production of structural proteins and virion assembly. Examples of late genes include genes coding for structural proteins (such as BcLF1 and BNRF1), glycoproteins (such as BLLF1 and BXLF2), and viral interleukin-10 (BCRF1) [65].
3. Overview of EBV Latency
Latent infections can establish one of four programs of gene expression: type 0, type I, type II, or type III (summarized in Figure 1). The proteins expressed play important roles in maintaining latency. A number of factors determining which latency program is ultimately expressed have been identified. For example, expression from the C promoter (cP) is essential in establishing type III latency. In type I latency, the cP is silent while the Q promoter (Qp) becomes active. An analysis of the three-dimensional structure of chromatin showed that the latent origin of replication (oriP), a section of the EBV chromosome important in replication and plasmid maintenance [67], is found near cP during type III latency, but near Qp during type I latency. CCCTC-binding factor (CTCF), which is a zinc finger protein important in creating DNA loops [68], has been implicated in modulating the association between oriP and either cP or Qp [69]. Furthermore, poly(ADP-ribose) polymerase I (PARP1) stabilizes CTCF binding and ensures that chromatin remains open during type III latency [70]. Histone H3 and H4 acetylation provide an additional layer of control over cP and Qp activation [71]. Establishment of type II latency occurs in the presence of IL-10, which can stimulate LMP1 production without concomitant stimulation of EBNA-2. This indicates that it may play a role in the establishment of type II latency [72]. EBV-infected cells expressing other types of latency can be converted to type II latency by IL-21, which stimulates latent membrane protein (LMP) 1 but not Epstein-Barr nuclear antigen (EBNA)-2 in type I latency. It inhibits cP and LMP2A mRNA while upregulating LMP1 mRNA in type III latency [73]. Type 0 latency is seen in B cells that are not undergoing active division [35].
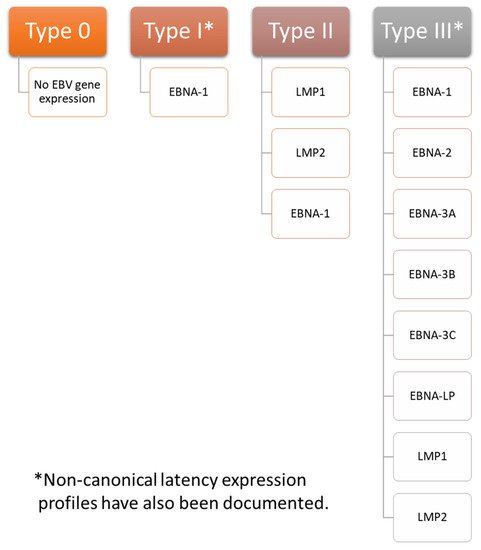
Figure 1. EBV Latency Types. EBV has four gene expression profiles during latent infection: type 0, type I, type II, and type III. In type 0 latency EBV, few, if any, proteins are expressed. Epstein-Barr nuclear antigen (EBNA)-1 is the only protein expressed during type I latency. Latent membrane protein (LMP) 1, LMP2, and EBNA-1 are all expressed during type II latency. All genes associated with latency are expressed in type III latency, including EBNA-1, EBNA-2, EBNA-3A, EBNA-3B, EBNA-3C, EBNA-LP (leader protein), LMP1, and LMP2. These proteins play important roles in maintaining latency. Notably, non-canonical latency expression profiles have also been documented. For example, infected cells expressing a type I latency profile may also express LMP1 or LMP2A, and infected cells expressing a type III latency profile may not express EBNA-2.
Few, if any, proteins are expressed during type 0 latency. EBNA-1 is the primary viral gene expressed in type I latency, although other latency-associated proteins are occasionally expressed as well. Infected cells exhibiting a type II latency pattern express LMP1 and 2 in addition to EBNA-1. Infected cells with a type III expression profile express all latency associated genes, including EBNA-1, EBNA-2, EBNA-3A, EBNA-3B, EBNA-3C, EBNA-LP (leader protein), LMP1, and LMP2 [35][74][75]. Of note, non-canonical patterns of EBV expression exist. For example, cells exhibiting a type I latency program may also express LMP1 or LMP2A [76], and cells with a type III latency expression profile may not express EBNA-2 [77].
Type I latency is associated with Burkitt lymphoma and can be modeled in Akata, Mutu, AG876, GC1, and YCCEL1 EBV strains. Type II latency can be found in nasopharyngeal carcinoma, Hodgkin lymphoma associated with EBV, and T cell lymphoma and can be modeled with the C666-1 EBV strain. Type III latency is seen in lymphoblastoid cell lines. It can be modeled with the B95-8, Raji, GD1, and GD2 EBV strains [35][74].
EBNA-1 is expressed in every lytic expression program except type 0. It contributes to the stability of the latent EBV episome [78], which is the EBV genome conformation most commonly assumed during latency [79], by playing roles in episomal replication and mitotic segregation. EBNA-1 activates other latent genes that lead to B cell immortalization [78] and tethers the viral episome to the host genome [80]. EBNA-2 is a transcription factor that interacts with DNA through the adaptor proteins, C-promoter binding factor (CBF) 1 and PU.1. It stimulates the C promoter as well as the promoters for LMP1, LMP2A, and LMP2B [81]. Of note, the C promoter is important in EBNA gene expression [82]. EBNA-2 is also essential for B cell immortalization [83]. The best characterized function of EBNA-LP is as a coactivator of EBNA-2 [81][84], but it has also been implicated in modulating other cellular and viral functions, such as apoptosis and cell survival [85][86]. It has been shown to help recruit transcription factors to the viral genome [86]. The EBNA-3 proteins are transcription regulators, with EBNA-3A and EBNA-3C acting as oncogenes and EBNA-3B acting as a tumor suppressor [87]. While EBNA-3B is nonessential for B cell immortalization [88], EBNA-3A and EBNA-3C are essential [89]. LMP1 is best known for its role in B cell immortalization, but it is also involved in a myriad of other viral functions, such as cell contact/migration, immunomodulation, changes in gene and miRNA expression, and stimulation of tumor invasion [90]. LMP2 has two isoforms, LMP2A and LMP2B. Together, these proteins are involved in transformation, proliferation, migration, and latency [91]. The two isoforms have conflicting roles in B cell receptor signaling, with LMP2A inhibiting B cell receptor signal transduction when expressed alone. Co-expression with LMP2B restored normal signal transduction [92]. LMP2A is also essential in generating surrogate cell survival signals [76][93].
In addition to the above proteins, EBV generates a host of RNAs during latency, including BamHI fragment A rightward transcripts (BARTs) and Epstein-Barr virus-encoded small RNAs (EBER). These RNAs play many essential roles in latently infected cells, including but not limited to contributions to cell survival, immunoevasion, cell proliferation, immunomodulation, malignant transformation, and maintaining viral latency [94][95][96][97][98]. Interestingly, EBER deletions have been shown to have no effect on establishing transformed lymphoblastoid cell lines [99], and it has been proposed that they act as a backup for LMP1 [100]. By contrast, BARTs have been shown to play a role in latency [101][102].
Establishment and Maintenance of Latency in EBV Infection
B cell immortalization is strongly associated with latency [103]. EBV has been shown to stimulate reactive oxygen species (ROS) production in cells [104][105]. This oxidative stress is required to immortalize B-cells. In fact, Chen, Kamranvar, and Masucci showed that the addition of ROS scavengers, such as N-acetylcysteine amide (NACA) and reduced glutathione, to infected B cells significantly inhibited their proliferation. No such adverse effect on cell growth was noted when ROS scavengers were added to mitogen-stimulated B cells. The authors further found that ROS production was necessary for normal expression of LMP1 and that ROS scavengers decreased signal transducer and activator of transcription 3 (STAT3) phosphorylation [106]. B cells derived from patients with a STAT3 negative mutation have previously been shown to resist EBV-induced immortalization, indicating the importance of STAT3 phosphorylation and activation in B cell transformation [107].
The early lytic cycle gene BHLF1 was recently shown to play a role in establishing latent infection and in B cell immortalization. Importantly, BHLF1 protein expression required the presence of the post-transcriptional regulator protein SM, which is only expressed during the lytic cycle. This requirement indicates that the latency-associated functions of BHLF1 are mediated by long noncoding RNA (lncRNA) rather than by the protein itself. While BL2 cells infected with both wild-type EBV and EBV carrying deletions of the BHLF1 open reading frame (ORF) and 5′ promoter region both established type III latency, cells infected with the mutant strain transitioned to a type I latency program by two months post infection. Cells infected by wild-type virus continued to express type III latency genes. This trend was also observed with deletions targeting the BHLF1 ORF alone, although the cells infected with this strain remained in type III latency for slightly longer than those infected with EBV containing deletions to both the BHLF1 ORF and 5′ promoter region. In addition, B cells from three out of four donors infected with either deletion demonstrated impaired B cell immortalization when compared to wild-type EBV. Cells from one donor were transformed equally effectively regardless of which of the three viruses were used to infect the cells, although the authors noted that these B cells appeared to be more sensitive to transformation than B cells from other donors [108]. Notably, the above experiments on B cell transformation were performed in vitro. There may be additional factors involved in in vivo transformation.
CRISPR screening recently identified MYC, an important cellular protein with many functions (reviewed in [109]), as a key regulator of EBV latency. Induction of lytic infection in Burkitt lymphoma cells was correlated with decreased MYC expression. MYC depletion resulted in increased expression of BZLF1 and BMRF1 in Akata and P3HR-1 cells, while eliminating MYC caused an increase in the expression of the late protein gp350 in Burkitt lymphoma cells. RNA sequencing analysis demonstrated that MYC depletion resulted in the upregulation of 77 genes associated with the EBV lytic replication cycle. There was a corresponding increase in the EBV genome copy number. MYC knockdown in lymphoblastoid B cells, which have a type III latent gene expression profile, also induced BZLF1 and BMRF1 expression. BMRF1 was not as strongly stimulated in lymphoblastoid cells, which the authors postulated may have been due to tet methylcytosine dioxygenase 2 (TET2) demethylase-mediated inhibition of BZLF’s ability to stimulate early gene expression. gp350 expression was abrogated in BZLF1 knockout cells, indicating that MYC acts on BZLF1 to promote latency. Subsequent experiments indicated that MYC acts on the BZLF1 promoter and interacts with EBV DNA near both oriLyts (origin of lytic replication). Chromatin conformation capture (3C) assay showed interactions between the BZLF1 promoter and the oriLyt T6 R primer as well as interactions between BZLF1 and the T10 primer of the TR region. These interactions did not occur in the presence of MYC overexpression. The authors proposed that loss of MYC results in oriLyt and TR-DNA looping to the BZLF1 promoter to trigger lytic replication [110].
The same set of experiments demonstrated that factors associated with MYC influence EBV latency. For example, depleting the cohesin structural maintenance of chromosomes 1A (SMC1A) also stimulated 77 EBV genes associated with the lytic cycle and increased the EBV genome copy number. Consistent with these results, MYC mRNA expression was significantly decreased. Likewise, the facilitated chromatin transcription (FACT) complex, which previously was shown to interact with MYC [111], impacts MYC expression. CRISPR targeting of SUPT16H, a FACT subunit, reduced MYC expression by 65% and stimulated 67 lytic genes. Additionally, SUPT16H and SSRP1, another FACT subunit, are both upregulated by EBV during initial infection. Other proteins in which the CRISPR knockout induced lytic gene expression included STAGA, GCN5 histone acetyltransferase/lysine acetyltransferase 2B (PCAF), and Mediator, which is recruited by STAGA and MYC [110].
EBV is capable of manipulating C-X-C motif chemokine receptor 4 (CXCR4) expression to maintain latency in EBV-related gastric carcinoma. EBV was shown to increase CXCR4 expression by activating the AKT/PI3K pathway. Notably, LMP2A was found to increase CXCR4 expression, and BZLF1 expression increased in AGS cells treated with siRNA targeting CXCR4. CXCR4 decreased the viral copy number and stimulated LMP2A and EBNA-1. These results indicate that CXCR4 is necessary for EBV latency [112].
Small ubiquitin-related modifier (SUMO) modification is a reversible physiologic process involved in regulating transcription, remodeling chromatin, and responding to hypoxic stress [113][114]. Like so many other cellular processes, it has been subverted for use by EBV [113]. EBV has three separate potential SUMO interaction motifs (SIMs) called SIM1, SIM2, and SIM3, of which SIM2 and SIM3 were shown to be important in EBNA-1’s ability to bind to His-tagged SUMO1 and SUMO2 proteins. SIM2 deletion inhibited EBNA-1 dimerization, while SIM3 deletion inhibited poly-SUMO2 modification of EBNA-1. Chromatin immunoprecipitation (ChIP) assays demonstrated that mutations in both SIM2 and SIM3 impaired the binding of EBNA-1 to oriP chromosomal DNA. Cells infected with EBV containing SIM3 or K477R (a SUMOylation site) mutations as well as SIM2, SIM3, and EBNA-1 with glycine/alanine deletions demonstrated impaired oriP mini genome maintenance. Furthermore, SIM3 deletion and K477R mutation stimulated the transcription of BZLF1, an immediate early protein important in lytic gene expression [115], while deletions in SIM2 or SIM3 caused increased transcriptional activity of the BZLF1 promoter. Immunoblot analysis demonstrated that SUMO1-modified proteins increased in frequency in the presence of EBNA-1, while SUMO2-modified proteins decreased in frequency. Notably, SUMO2-associated proteins targeted by EBNA-1 were primarily associated with the proteasome regulatory complex and the ubiquitin-dependent Cullin-RING E3 ligase. This indicates that EBNA-1 may target proteins with SUMO2 modifications for degradation. Pathways affected include those involved in the viral life cycle, gene transcription, gene expression, and mRNA metabolic processes. Those affecting DNA and RNA binding were particularly prominent in affected mRNA metabolic process pathways. In addition, proteins associated with EBNA-1 SIM sites were mainly associated with DNA and RNA binding, gene transcription and expression, and other proteins involved in proteasome-mediated degradation. Lastly, the authors showed that hypoxia-induced EBV reactivation stimulated an increase in SUMO1-modified STIP1 homology and U-box containing protein 1 (STUB1) and tripartite motif containing 28 (KAP1), while the SUMO2 modified forms decreased. Hypoxia stimulated the preferential association of EBNA-1 with SUMO2 over SUMO1. It also downregulated the association between EBNA-1 and SUMO2-modified KAP1, which, along with ubiquitin specific peptidase 7 (USP7), make up a SUMO2-modified complex. STUB1 inhibition decreased the EBV genome copy number, while USP7 knockdown had the opposite effect. KAP1, USP7, and STUB1 knockdown cells exposed to hypoxic conditions had higher BZLF1 levels than the control [116].
Paired box (PAX) 5 is a cellular oncogene involved in activating genes that promote differentiation into B cells and in inhibiting genes that promote differentiation into other cell types [117]. More recently, it has been shown to physically interact with EBNA-1. Expression of a short hairpin RNA targeting the 3′ untranslated region of PAX5 nearly eliminated the activity of the oriP-Luc reporter plasmid, which is an oriP-SV40-Luciferase expression vector dependent on EBNA-1 expression [118]. This defect was corrected in the presence of plasmid-generated Flag epitope-tagged PAX5 (FPAX5). PAX5 depletion resulted in a 70%–95% decrease in EBNA-1 enrichment at oriP and a 50%–95% decrease in nucleolin (NCL, an EBNA-1 associated protein) enrichment at TR-DNA, as measured by ChIP assays. This is consistent with a model in which PAX5 is necessary for EBNA-1 to localize to EBV oriP or TR-DNA. It was also shown that PAX5 associates with the transcription enhancers p300 and histone 3 lysine 4 trimethyl (H3K4me3). PAX5 knockdown resulted in the dissociation of p300 from oriP DNA as well as the dissociation of both p300 and H3K4me3 from TR-DNA. EBNA-1 dissociated from both oriP DNA and TR-DNA. Lastly, the authors showed that PAX5 knockdown reduced the EBV genome copy number, a finding that was reversed in the presence of transfected FPAX5 [119].
Subverting histone chaperone proteins is another mechanism by which EBV maintains latency. For example, the chromatin assembly factor (CAF) 1 complex supplies histone 3 and histone 4 dimers to replication forks. Depletion of any of CAF’s three subunits (CHAF1A, CHAF1B, and RBBP4) induced the expression of BZLF1 (an immediate early gene) and BMRF1 (an early gene) as well as numerous transcripts associated with lytic infection. These results were seen in multiple Burkitt’s lymphoma cell lines. In addition, it has been shown that inhibiting EBNA-2 resulted in downregulation of CAF1 subunit mRNA expression. Downregulating histone loader histone regulatory homologue A (HIRA), which loads H3.3 and H4 complexes onto DNA, had a similar result in that it increased BZLF1 and BMRF1 expression. Unlike CHAF1A and CHAF1B, there was no decrease in HIRA mRNA levels. Additionally, targeting alpha thalassemia/mental retardation syndrome X-linked chromatin remodeler (ATRX) and death domain-associated protein (DAXX), which load histone 3.3, with CRISPR stimulated BZLF1 and BMRF1 expression. This indicates that EBV subverts multiple histone loaders to maintain latent infection. Interestingly, reduction in CHAF1B levels decreased the concentration of histone 3.1 and histone 3.3 at the promoters for BZLF1 and BLLF1, and CHAF1B single guide RNA (sgCHAF1B) reduced histone 3.1 presence at oriLyt L and oriLyt R, which are important in initiating lytic gene expression. Levels of histone 3.1 and histone 3.3 were both increased by two days post infection, indicating that these histones are transferred to incoming EBV episomes. The same study showed that CHAF1B knockdown decreased histone 3 lysine 9 trimethyl (H3K9me3) at the promoters for BZLF1, BLLF1, oriLyt R, and oriLyt L, and that sgCHAF1B reduced histone 3 lysine 27 trimethyl (H3K27me3) occupancy at these sites in Akata cells [120]. Both H3K9me3 and H3K27me3 are repressive histone modifications [121].
Recent research has demonstrated the importance of miRNA in maintaining latency. Screening identified eight miRNAs (BHRF1-2, BART1, BART2, BART8, BART11, BART18, BART9, and BART17) whose expression diminished nuclear factor kappa B (NF-κB) expression, which was used as a proxy for B cell receptor activation. In addition, BHRF1-1, BHRF1-2, BART14, and BART18 expression significantly reduced AP-1 signaling. B cell receptor engagement leads to the activation of the transcription factor AP-1 via the GTPase Ral [122]. Furthermore, many targets of EBV mRNAs are involved in B cell receptor signaling, such as growth factor receptor-bound protein 2 (GRB2), SOS Ras/Rac guanine nucleotide exchange factor 1 (SOS1), Ras-related C3 botulinum toxin substrate 1 (RAC1), and Ikk-B. shRNA targeting GRB2, SOS1, or RAC1 resulted in a sharp downregulation of NF-κB. Intriguingly, miR-BHRF1-2-5p targets all three of these proteins. Indeed, miR-BHRF1-2-5P inhibition stunted the growth of both lymphoblastoid and EBV-associated diffuse large B cell lymphoma cell lines [123]. A summary of mechanisms involved in EBV latency can be found in Table 1.
Table 1. Mechanisms Involved in EBV Latency.
| Mechanism | Effect | References |
|---|---|---|
| ROS Expression | B cell immortalization | [104][105][106] |
| - | Required for normal LMP1 expression | [106] |
| - | STAT3 phosphorylation | [106][107] |
| BHLF1 | Maintenance of type III latency | [108] |
| PAX5 | EBNA-1 localization to oriP and TR-DNA | [119] |
| - | Association of transcription enhancers from oriP and TR-DNA | [119] |
| CAF1 | Inhibits lytic gene expression and increases histone presence at multiple points on the EBV genome. | [120] |
| HIRA | Histone loader involved in maintaining latency | [120] |
| ATRX | Histone loader involved in maintaining latency | [120] |
| DAXX | Histone loader involved in maintaining latency | [120] |
| MYC | Acts on BZLF1 promoter to prevent oriLyt and TR-DNA from looping | [110] |
| SMC1A | Contributes to latency by promoting MYC expression | [110] |
| Facilitated Chromatic Transcription Complex | Contributes to latency by promoting MYC expression | [110] |
| CXCR4 | Maintenance of latency; stimulates LMP2A and EBNA-1 | [112] |
| SUMOylation/SIM-interacting motifs | Facilitates oriP mini genome maintenance and the binding of EBNA-1 to His-tagged SUMO1 and SUMO2 proteins | [116] |
| - | EBNA-1 targets proteins with SUMO2 modifications for degradation | [116] |
| - | Inhibits BZLF1 expression | [116] |
| miRNAs | Inhibition of B cell receptor activation by diminishing NF-κB and/or AP-1 signaling | [123] |
References
- Dunmire, S.K.; Verghese, P.S.; Balfour, H.H., Jr. Primary Epstein-Barr virus infection. J. Clin. Virol. 2018, 102, 84–92.
- Fugl, A.; Andersen, C.L. Epstein-Barr virus and its association with disease—A review of relevance to general practice. BMC Fam. Pract. 2019, 20, 62.
- Womack, J.; Jimenez, M. Common questions about infectious mononucleosis. Am. Fam. Phys. 2015, 91, 372–376.
- Kerr, J.R. Epstein-Barr virus (EBV) reactivation and therapeutic inhibitors. J. Clin. Pathol. 2019, 72, 651–658.
- Dunmire, S.K.; Hogquist, K.A.; Balfour, H.H. Infectious Mononucleosis. Curr. Top. Microbiol. Immunol. 2015, 390, 211–240.
- Houen, G.; Trier, N.H. Epstein-Barr Virus and Systemic Autoimmune Diseases. Front. Immunol. 2020, 11, 587380.
- Pedersen, M.; Asprusten, T.T.; Godang, K.; Leegaard, T.M.; Osnes, L.T.; Skovlund, E.; Tjade, T.; Oie, M.G.; Wyller, V.B.B. Predictors of chronic fatigue in adolescents six months after acute Epstein-Barr virus infection: A prospective cohort study. Brain Behav. Immun. 2019, 75, 94–100.
- Nagata, K.; Hara, S.; Nakayama, Y.; Higaki, K.; Sugihara, H.; Kuwamoto, S.; Matsushita, M.; Kato, M.; Tanio, S.; Ishiguro, K.; et al. Epstein-Barr Virus Lytic Reactivation Induces IgG4 Production by Host B Lymphocytes in Graves’ Disease Patients and Controls: A Subset of Graves’ Disease Is an IgG4-Related Disease-Like Condition. Viral Immunol. 2018, 31, 540–547.
- Al Hamed, R.; Bazarbachi, A.H.; Mohty, M. Epstein-Barr virus-related post-transplant lymphoproliferative disease (EBV-PTLD) in the setting of allogeneic stem cell transplantation: A comprehensive review from pathogenesis to forthcoming treatment modalities. Bone Marrow Transpl. 2020, 55, 25–39.
- Houen, G.; Trier, N.H.; Frederiksen, J.L. Epstein-Barr Virus and Multiple Sclerosis. Front. Immunol. 2020, 11, 587078.
- Ou, Y.N.; Zhu, J.X.; Hou, X.H.; Shen, X.N.; Xu, W.; Dong, Q.; Tan, L.; Yu, J.T. Associations of Infectious Agents with Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2020, 75, 299–309.
- Robinson, T.J.; Glenn, M.S.; Temple, R.W.; Wyatt, D.; Connolly, J.H. Encephalitis and cerebellar ataxia associated with Epstein-Barr virus infections. Ulster Med. J. 1980, 49, 158–164.
- Abul-Kasim, K.; Palm, L.; Maly, P.; Sundgren, P.C. The neuroanatomic localization of Epstein-Barr virus encephalitis may be a predictive factor for its clinical outcome: A case report and review of 100 cases in 28 reports. J. Child. Neurol. 2009, 24, 720–726.
- Greenspan, J.S.; Greenspan, D.; Lennette, E.T.; Abrams, D.I.; Conant, M.A.; Petersen, V.; Freese, U.K. Replication of Epstein-Barr virus within the epithelial cells of oral "hairy" leukoplakia, an AIDS-associated lesion. N. Engl. J. Med. 1985, 313, 1564–1571.
- Ko, Y.H. EBV and human cancer. Exp. Mol. Med. 2015, 47, e130.
- Neparidze, N.; Lacy, J. Malignancies associated with epstein-barr virus: Pathobiology, clinical features, and evolving treatments. Clin. Adv. Hematol. Oncol. 2014, 12, 358–371.
- Tsao, S.W.; Tsang, C.M.; To, K.F.; Lo, K.W. The role of Epstein-Barr virus in epithelial malignancies. J. Pathol. 2015, 235, 323–333.
- Utsuki, S.; Oka, H.; Miyajima, Y.; Kijima, C.; Yasui, Y.; Fujii, K. Epstein-Barr virus (EBV)-associated primary central nervous system lymphoma: Is incidence of EBV expression associated with median survival time? Brain Tumor Pathol. 2011, 28, 145–149.
- Khan, G.; Hashim, M.J. Global burden of deaths from Epstein-Barr virus attributable malignancies 1990–2010. Infect. Agent Cancer 2014, 9, 38.
- Khan, G.; Fitzmaurice, C.; Naghavi, M.; Ahmed, L.A. Global and regional incidence, mortality and disability-adjusted life-years for Epstein-Barr virus-attributable malignancies, 1990–2017. BMJ Open 2020, 10, e037505.
- Drosu, N.C.; Edelman, E.R.; Housman, D.E. Tenofovir prodrugs potently inhibit Epstein-Barr virus lytic DNA replication by targeting the viral DNA polymerase. Proc. Natl. Acad. Sci. USA 2020, 117, 12368–12374.
- Cohen, J.I. Vaccine Development for Epstein-Barr Virus. Adv. Exp. Med. Biol. 2018, 1045, 477–493.
- Pellett, P.E.; Roizman, B. Herpesviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1802–1822.
- Machon, C.; Fabrega-Ferrer, M.; Zhou, D.; Cuervo, A.; Carrascosa, J.L.; Stuart, D.I.; Coll, M. Atomic structure of the Epstein-Barr virus portal. Nat. Commun. 2019, 10, 3891.
- Liu, X.; Cohen, J.I. Epstein-Barr Virus (EBV) Tegument Protein BGLF2 Promotes EBV Reactivation through Activation of the p38 Mitogen-Activated Protein Kinase. J. Virol. 2016, 90, 1129–1138.
- He, H.P.; Luo, M.; Cao, Y.L.; Lin, Y.X.; Zhang, H.; Zhang, X.; Ou, J.Y.; Yu, B.; Chen, X.; Xu, M.; et al. Structure of Epstein-Barr virus tegument protein complex BBRF2-BSRF1 reveals its potential role in viral envelopment. Nat. Commun. 2020, 11, 5405.
- van Gent, M.; Braem, S.G.; de Jong, A.; Delagic, N.; Peeters, J.G.; Boer, I.G.; Moynagh, P.N.; Kremmer, E.; Wiertz, E.J.; Ovaa, H.; et al. Epstein-Barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with toll-like receptor signaling. PLoS Pathog. 2014, 10, e1003960.
- Chen, T.; Wang, Y.; Xu, Z.; Zou, X.; Wang, P.; Ou, X.; Li, Y.; Peng, T.; Chen, D.; Li, M.; et al. Epstein-Barr virus tegument protein BGLF2 inhibits NF-kappaB activity by preventing p65 Ser536 phosphorylation. FASEB J. 2019, 33, 10563–10576.
- Hutt-Fletcher, L.M. EBV glycoproteins: Where are we now? Future Virol. 2015, 10, 1155–1162.
- Tosato, G.; Cohen, J.I. Generation of Epstein-Barr Virus (EBV)-immortalized B cell lines. Curr. Protoc. Immunol. 2007, 7, 22.
- Chen, J.; Longnecker, R. Epithelial cell infection by Epstein-Barr virus. FEMS Microbiol. Rev. 2019, 43, 674–683.
- Connolly, S.A.; Jackson, J.O.; Jardetzky, T.S.; Longnecker, R. Fusing structure and function: A structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 2011, 9, 369–381.
- Heldwein, E.E. gH/gL supercomplexes at early stages of herpesvirus entry. Curr. Opin. Virol. 2016, 18, 1–8.
- Young, K.A.; Chen, X.S.; Holers, V.M.; Hannan, J.P. Isolating the Epstein-Barr virus gp350/220 binding site on complement receptor type 2 (CR2/CD21). J. Biol. Chem. 2007, 282, 36614–36625.
- Longnecker, R.M.; Kieff, E.; Cohen, J.I. Epstein-Barr Virus. In Field’s Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1898–1959.
- Wang, H.B.; Zhang, H.; Zhang, J.P.; Li, Y.; Zhao, B.; Feng, G.K.; Du, Y.; Xiong, D.; Zhong, Q.; Liu, W.L.; et al. Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat. Commun. 2015, 6, 6240.
- Zhang, H.; Li, Y.; Wang, H.B.; Zhang, A.; Chen, M.L.; Fang, Z.X.; Dong, X.D.; Li, S.B.; Du, Y.; Xiong, D.; et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein-Barr virus entry. Nat. Microbiol. 2018, 3, 1–8.
- Xiong, D.; Du, Y.; Wang, H.B.; Zhao, B.; Zhang, H.; Li, Y.; Hu, L.J.; Cao, J.Y.; Zhong, Q.; Liu, W.L.; et al. Nonmuscle myosin heavy chain IIA mediates Epstein-Barr virus infection of nasopharyngeal epithelial cells. Proc. Natl. Acad. Sci. USA 2015, 112, 11036–11041.
- Fingeroth, J.D.; Diamond, M.E.; Sage, D.R.; Hayman, J.; Yates, J.L. CD21-Dependent infection of an epithelial cell line, 293, by Epstein-Barr virus. J. Virol. 1999, 73, 2115–2125.
- Turk, S.M.; Jiang, R.; Chesnokova, L.S.; Hutt-Fletcher, L.M. Antibodies to gp350/220 enhance the ability of Epstein-Barr virus to infect epithelial cells. J. Virol. 2006, 80, 9628–9633.
- Janz, A.; Oezel, M.; Kurzeder, C.; Mautner, J.; Pich, D.; Kost, M.; Hammerschmidt, W.; Delecluse, H.J. Infectious Epstein-Barr virus lacking major glycoprotein BLLF1 (gp350/220) demonstrates the existence of additional viral ligands. J. Virol. 2000, 74, 10142–10152.
- Smith, N.A.; Coleman, C.B.; Gewurz, B.E.; Rochford, R. CD21 (Complement Receptor 2) Is the Receptor for Epstein-Barr Virus Entry into T Cells. J. Virol. 2020, 94, e00428-20.
- Lee, J.H.; Choi, J.; Ahn, Y.O.; Kim, T.M.; Heo, D.S. CD21-independent Epstein-Barr virus entry into NK cells. Cell Immunol. 2018, 327, 21–25.
- Tabiasco, J.; Vercellone, A.; Meggetto, F.; Hudrisier, D.; Brousset, P.; Fournie, J.J. Acquisition of viral receptor by NK cells through immunological synapse. J. Immunol. 2003, 170, 5993–5998.
- Mohl, B.S.; Chen, J.; Sathiyamoorthy, K.; Jardetzky, T.S.; Longnecker, R. Structural and Mechanistic Insights into the Tropism of Epstein-Barr Virus. Mol. Cells 2016, 39, 286–291.
- Shannon-Lowe, C.; Rowe, M. Epstein Barr virus entry; kissing and conjugation. Curr. Opin. Virol. 2014, 4, 78–84.
- Hui-Yuen, J.; McAllister, S.; Koganti, S.; Hill, E.; Bhaduri-McIntosh, S. Establishment of Epstein-Barr virus growth-transformed lymphoblastoid cell lines. J. Vis. Exp. 2011, 57, 3321.
- Jiang, R.; Kanamori, M.; Satoh, Y.; Fukuda, M.; Ikuta, K.; Murakami, M.; Sairenji, T. Contrasting effects of hydroxyurea on cell growth and reduction in Epstein-Barr virus genomes in EBV-infected epithelioid cell lines vs Burkitt’s lymphoma cell lines. J. Med. Virol. 2003, 70, 244–252.
- Edwards, R.H.; Dekroon, R.; Raab-Traub, N. Alterations in cellular expression in EBV infected epithelial cell lines and tumors. PLoS Pathog. 2019, 15, e1008071.
- Callard, R.E.; Lau, Y.L.; Shields, J.G.; Smith, S.H.; Cairns, J.; Flores-Romo, L.; Gordon, J. The marmoset B-lymphoblastoid cell line (B95-8) produces and responds to B-cell growth and differentiation factors: Role of shed CD23 (sCD23). Immunology 1988, 65, 379–384.
- Isaksson, A.; Berggren, M.; Ekeland-Sjoberg, K.; Samuelsson, T.; Ricksten, A. Cell specific internal translation efficiency of Epstein-Barr virus present in solid organ transplant patients. J. Med. Virol. 2007, 79, 573–581.
- Savard, M.; Belanger, C.; Tardif, M.; Gourde, P.; Flamand, L.; Gosselin, J. Infection of primary human monocytes by Epstein-Barr virus. J. Virol. 2000, 74, 2612–2619.
- Masy, E.; Adriaenssens, E.; Montpellier, C.; Crepieux, P.; Mougel, A.; Quatannens, B.; Goormachtigh, G.; Faumont, N.; Meggetto, F.; Auriault, C.; et al. Human monocytic cell lines transformed in vitro by Epstein-Barr virus display a type II latency and LMP-1-dependent proliferation. J. Virol. 2002, 76, 6460–6472.
- Jha, H.C.; Mehta, D.; Lu, J.; El-Naccache, D.; Shukla, S.K.; Kovacsics, C.; Kolson, D.; Robertson, E.S. Gammaherpesvirus Infection of Human Neuronal Cells. mBio 2015, 6, e01844-15.
- Tiwari, D.; Jakhmola, S.; Pathak, D.K.; Kumar, R.; Jha, H.C. Temporal In Vitro Raman Spectroscopy for Monitoring Replication Kinetics of Epstein-Barr Virus Infection in Glial Cells. ACS Omega 2020, 5, 29547–29560.
- Gruffat, H.; Marchione, R.; Manet, E. Herpesvirus Late Gene Expression: A Viral-Specific Pre-initiation Complex Is Key. Front. Microbiol. 2016, 7, 869.
- McKenzie, J.; El-Guindy, A. Epstein-Barr Virus Lytic Cycle Reactivation. Curr. Top. Microbiol. Immunol. 2015, 391, 237–261.
- Feederle, R.; Kost, M.; Baumann, M.; Janz, A.; Drouet, E.; Hammerschmidt, W.; Delecluse, H.J. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 2000, 19, 3080–3089.
- Ruvolo, V.; Wang, E.; Boyle, S.; Swaminathan, S. The Epstein-Barr virus nuclear protein SM is both a post-transcriptional inhibitor and activator of gene expression. Proc. Natl. Acad. Sci. USA 1998, 95, 8852–8857.
- Semmes, O.J.; Chen, L.; Sarisky, R.T.; Gao, Z.; Zhong, L.; Hayward, S.D. Mta has properties of an RNA export protein and increases cytoplasmic accumulation of Epstein-Barr virus replication gene mRNA. J. Virol. 1998, 72, 9526–9534.
- Tsurumi, T.; Daikoku, T.; Kurachi, R.; Nishiyama, Y. Functional interaction between Epstein-Barr virus DNA polymerase catalytic subunit and its accessory subunit in vitro. J. Virol. 1993, 67, 7648–7653.
- Strockbine, L.D.; Cohen, J.I.; Farrah, T.; Lyman, S.D.; Wagener, F.; DuBose, R.F.; Armitage, R.J.; Spriggs, M.K. The Epstein-Barr virus BARF1 gene encodes a novel, soluble colony-stimulating factor-1 receptor. J. Virol. 1998, 72, 4015–4021.
- Henderson, S.; Huen, D.; Rowe, M.; Dawson, C.; Johnson, G.; Rickinson, A. Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc. Natl. Acad. Sci. USA 1993, 90, 8479–8483.
- Summers, W.C.; Klein, G. Inhibition of Epstein-Barr virus DNA synthesis and late gene expression by phosphonoacetic acid. J. Virol. 1976, 18, 151–155.
- Young, L.S.; Arrand, J.R.; Murray, P.G. EBV gene expression and regulation. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007.
- Aubry, V.; Mure, F.; Mariame, B.; Deschamps, T.; Wyrwicz, L.S.; Manet, E.; Gruffat, H. Epstein-Barr virus late gene transcription depends on the assembly of a virus-specific preinitiation complex. J. Virol. 2014, 88, 12825–12838.
- Yates, J.L.; Camiolo, S.M.; Bashaw, J.M. The minimal replicator of Epstein-Barr virus oriP. J. Virol. 2000, 74, 4512–4522.
- Phillips, J.E.; Corces, V.G. CTCF: Master weaver of the genome. Cell 2009, 137, 1194–1211.
- Tempera, I.; Klichinsky, M.; Lieberman, P.M. EBV latency types adopt alternative chromatin conformations. PLoS Pathog. 2011, 7, e1002180.
- Lupey-Green, L.N.; Caruso, L.B.; Madzo, J.; Martin, K.A.; Tan, Y.; Hulse, M.; Tempera, I. PARP1 Stabilizes CTCF Binding and Chromatin Structure to Maintain Epstein-Barr Virus Latency Type. J. Virol. 2018, 92, e00755-18.
- Fejer, G.; Koroknai, A.; Banati, F.; Gyory, I.; Salamon, D.; Wolf, H.; Niller, H.H.; Minarovits, J. Latency type-specific distribution of epigenetic marks at the alternative promoters Cp and Qp of Epstein-Barr virus. J. Gen. Virol. 2008, 89, 1364–1370.
- Kis, L.L.; Takahara, M.; Nagy, N.; Klein, G.; Klein, E. IL-10 can induce the expression of EBV-encoded latent membrane protein-1 (LMP-1) in the absence of EBNA-2 in B lymphocytes and in Burkitt lymphoma- and NK lymphoma-derived cell lines. Blood 2006, 107, 2928–2935.
- Kis, L.L.; Salamon, D.; Persson, E.K.; Nagy, N.; Scheeren, F.A.; Spits, H.; Klein, G.; Klein, E. IL-21 imposes a type II EBV gene expression on type III and type I B cells by the repression of C- and activation of LMP-1-promoter. Proc. Natl. Acad. Sci. USA 2010, 107, 872–877.
- Choi, S.J.; Jung, S.W.; Huh, S.; Cho, H.; Kang, H. Phylogenetic comparison of Epstein-Barr virus genomes. J. Microbiol. 2018, 56, 525–533.
- Murata, T. Regulation of Epstein-Barr virus reactivation from latency. Microbiol. Immunol. 2014, 58, 307–317.
- Granai, M.; Mundo, L.; Akarca, A.U.; Siciliano, M.C.; Rizvi, H.; Mancini, V.; Onyango, N.; Nyagol, J.; Abinya, N.O.; Maha, I.; et al. Immune landscape in Burkitt lymphoma reveals M2-macrophage polarization and correlation between PD-L1 expression and non-canonical EBV latency program. Infect. Agent Cancer 2020, 15, 28.
- Touitou, R.; Arbach, H.; Cochet, C.; Feuillard, J.; Martin, A.; Raphael, M.; Joab, I. Heterogeneous Epstein-Barr virus latent gene expression in AIDS-associated lymphomas and in type I Burkitt’s lymphoma cell lines. J. Gen. Virol 2003, 84, 949–957.
- Frappier, L. Ebna1. Curr. Top. Microbiol. Immunol. 2015, 391, 3–34.
- Lieberman, P.M. Chromatin Structure of Epstein-Barr Virus Latent Episomes. Curr. Top. Microbiol. Immunol. 2015, 390, 71–102.
- Kim, K.D.; Tanizawa, H.; De Leo, A.; Vladimirova, O.; Kossenkov, A.; Lu, F.; Showe, L.C.; Noma, K.I.; Lieberman, P.M. Epigenetic specifications of host chromosome docking sites for latent Epstein-Barr virus. Nat. Commun. 2020, 11, 877.
- Kempkes, B.; Ling, P.D. EBNA2 and Its Coactivator EBNA-LP. Curr. Top. Microbiol. Immunol. 2015, 391, 35–59.
- Jin, X.W.; Speck, S.H. Identification of critical cis elements involved in mediating Epstein-Barr virus nuclear antigen 2-dependent activity of an enhancer located upstream of the viral BamHI C promoter. J. Virol. 1992, 66, 2846–2852.
- Hammerschmidt, W.; Sugden, B. Genetic analysis of immortalizing functions of Epstein-Barr virus in human B lymphocytes. Nature 1989, 340, 393–397.
- Harada, S.; Kieff, E. Epstein-Barr virus nuclear protein LP stimulates EBNA-2 acidic domain-mediated transcriptional activation. J. Virol. 1997, 71, 6611–6618.
- Matsuda, G.; Nakajima, K.; Kawaguchi, Y.; Yamanashi, Y.; Hirai, K. Epstein-Barr virus (EBV) nuclear antigen leader protein (EBNA-LP) forms complexes with a cellular anti-apoptosis protein Bcl-2 or its EBV counterpart BHRF1 through HS1-associated protein X-1. Microbiol. Immunol. 2003, 47, 91–99.
- Szymula, A.; Palermo, R.D.; Bayoumy, A.; Groves, I.J.; Ba Abdullah, M.; Holder, B.; White, R.E. Epstein-Barr virus nuclear antigen EBNA-LP is essential for transforming naive B cells, and facilitates recruitment of transcription factors to the viral genome. PLoS Pathog. 2018, 14, e1006890.
- Styles, C.T.; Paschos, K.; White, R.E.; Farrell, P.J. The Cooperative Functions of the EBNA3 Proteins Are Central to EBV Persistence and Latency. Pathogens 2018, 7, 31.
- Chen, A.; Divisconte, M.; Jiang, X.; Quink, C.; Wang, F. Epstein-Barr virus with the latent infection nuclear antigen 3B completely deleted is still competent for B-cell growth transformation in vitro. J. Virol. 2005, 79, 4506–4509.
- Tomkinson, B.; Robertson, E.; Kieff, E. Epstein-Barr virus nuclear proteins EBNA-3A and EBNA-3C are essential for B-lymphocyte growth transformation. J. Virol. 1993, 67, 2014–2025.
- Kieser, A.; Sterz, K.R. The Latent Membrane Protein 1 (LMP1). Curr. Top. Microbiol. Immunol. 2015, 391, 119–149.
- Cen, O.; Longnecker, R. Latent Membrane Protein 2 (LMP2). Curr. Top. Microbiol. Immunol. 2015, 391, 151–180.
- Rovedo, M.; Longnecker, R. Epstein-barr virus latent membrane protein 2B (LMP2B) modulates LMP2A activity. J. Virol. 2007, 81, 84–94.
- Merchant, M.; Caldwell, R.G.; Longnecker, R. The LMP2A ITAM is essential for providing B cells with development and survival signals in vivo. J. Virol. 2000, 74, 9115–9124.
- Kang, D.; Skalsky, R.L.; Cullen, B.R. EBV BART MicroRNAs Target Multiple Pro-apoptotic Cellular Genes to Promote Epithelial Cell Survival. PLoS Pathog. 2015, 11, e1004979.
- Wang, Y.; Guo, Z.; Shu, Y.; Zhou, H.; Wang, H.; Zhang, W. BART miRNAs: An unimaginable force in the development of nasopharyngeal carcinoma. Eur. J. Cancer Prev. 2017, 26, 144–150.
- Takada, K. Role of EBER and BARF1 in nasopharyngeal carcinoma (NPC) tumorigenesis. Semin. Cancer Biol. 2012, 22, 162–165.
- Iwakiri, D. Epstein-Barr Virus-Encoded RNAs: Key Molecules in Viral Pathogenesis. Cancers 2014, 6, 1615–1630.
- Dong, M.; Chen, J.N.; Huang, J.T.; Gong, L.P.; Shao, C.K. The roles of EBV-encoded microRNAs in EBV-associated tumors. Crit. Rev. Oncol. Hematol. 2019, 135, 30–38.
- Swaminathan, S.; Tomkinson, B.; Kieff, E. Recombinant Epstein-Barr virus with small RNA (EBER) genes deleted transforms lymphocytes and replicates in vitro. Proc. Natl. Acad. Sci. USA 1991, 88, 1546–1550.
- Herbert, K.M.; Pimienta, G. Consideration of Epstein-Barr Virus-Encoded Noncoding RNAs EBER1 and EBER2 as a Functional Backup of Viral Oncoprotein Latent Membrane Protein 1. mBio 2016, 7, e01926-15.
- Verhoeven, R.J.A.; Tong, S.; Mok, B.W.; Liu, J.; He, S.; Zong, J.; Chen, Y.; Tsao, S.W.; Lung, M.L.; Chen, H. Epstein-Barr Virus BART Long Non-coding RNAs Function as Epigenetic Modulators in Nasopharyngeal Carcinoma. Front. Oncol. 2019, 9, 1120.
- De Re, V.; Caggiari, L.; De Zorzi, M.; Fanotto, V.; Miolo, G.; Puglisi, F.; Cannizzaro, R.; Canzonieri, V.; Steffan, A.; Farruggia, P.; et al. Epstein-Barr virus BART microRNAs in EBV- associated Hodgkin lymphoma and gastric cancer. Infect. Agent Cancer 2020, 15, 42.
- Leibold, W.; Flanagan, T.D.; Menezes, J.; Klein, G. Induction of Epstein-Barr virus-associated nuclear antigen during in vitro transformation of human lymphoid cells. J. Natl. Cancer Inst. 1975, 54, 65–68.
- Gruhne, B.; Sompallae, R.; Marescotti, D.; Kamranvar, S.A.; Gastaldello, S.; Masucci, M.G. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2009, 106, 2313–2318.
- Hu, J.; Li, H.; Luo, X.; Li, Y.; Bode, A.; Cao, Y. The role of oxidative stress in EBV lytic reactivation, radioresistance and the potential preventive and therapeutic implications. Int. J. Cancer 2017, 141, 1722–1729.
- Chen, X.; Kamranvar, S.A.; Masucci, M.G. Oxidative stress enables Epstein-Barr virus-induced B-cell transformation by posttranscriptional regulation of viral and cellular growth-promoting factors. Oncogene 2016, 35, 3807–3816.
- Koganti, S.; de la Paz, A.; Freeman, A.F.; Bhaduri-McIntosh, S. B lymphocytes from patients with a hypomorphic mutation in STAT3 resist Epstein-Barr virus-driven cell proliferation. J. Virol. 2014, 88, 516–524.
- Yetming, K.D.; Lupey-Green, L.N.; Biryukov, S.; Hughes, D.J.; Marendy, E.M.; Miranda, J.L.; Sample, J.T. The BHLF1 Locus of Epstein-Barr Virus Contributes to Viral Latency and B-Cell Immortalization. J. Virol. 2020, 94, e01215-20.
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357.
- Guo, R.; Jiang, C.; Zhang, Y.; Govande, A.; Trudeau, S.J.; Chen, F.; Fry, C.J.; Puri, R.; Wolinsky, E.; Schineller, M.; et al. MYC Controls the Epstein-Barr Virus Lytic Switch. Mol. Cell 2020, 78, 653–669.
- Carter, D.R.; Murray, J.; Cheung, B.B.; Gamble, L.; Koach, J.; Tsang, J.; Sutton, S.; Kalla, H.; Syed, S.; Gifford, A.J.; et al. Therapeutic targeting of the MYC signal by inhibition of histone chaperone FACT in neuroblastoma. Sci. Transl. Med. 2015, 7, 312ra176.
- Wang, W.; Zhang, Y.; Liu, W.; Zhang, X.; Xiao, H.; Zhao, M.; Luo, B. CXCR4 induces cell autophagy and maintains EBV latent infection in EBVaGC. Theranostics 2020, 10, 11549–11561.
- Chang, P.C.; Campbell, M.; Robertson, E.S. Human Oncogenic Herpesvirus and Post-translational Modifications—Phosphorylation and SUMOylation. Front. Microbiol. 2016, 7, 962.
- Cheng, J.; Kang, X.; Zhang, S.; Yeh, E.T. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell 2007, 131, 584–595.
- Chen, C.; Li, D.; Guo, N. Regulation of cellular and viral protein expression by the Epstein-Barr virus transcriptional regulator Zta: Implications for therapy of EBV associated tumors. Cancer Biol. Ther. 2009, 8, 987–995.
- Wang, Y.; Du, S.; Zhu, C.; Wang, C.; Yu, N.; Lin, Z.; Gan, J.; Guo, Y.; Huang, X.; He, Y.; et al. STUB1 is targeted by the SUMO-interacting motif of EBNA1 to maintain Epstein-Barr Virus latency. PLoS Pathog. 2020, 16, e1008447.
- O’Brien, P.; Morin, P., Jr.; Ouellette, R.J.; Robichaud, G.A. The Pax-5 gene: A pluripotent regulator of B-cell differentiation and cancer disease. Cancer Res. 2011, 71, 7345–7350.
- Chen, Y.L.; Tsai, H.L.; Peng, C.W. EGCG debilitates the persistence of EBV latency by reducing the DNA binding potency of nuclear antigen 1. Biochem. Biophys. Res. Commun. 2012, 417, 1093–1099.
- Liu, C.D.; Lee, H.L.; Peng, C.W. B Cell-Specific Transcription Activator PAX5 Recruits p300 To Support EBNA1-Driven Transcription. J. Virol. 2020, 94, e02028-19.
- Zhang, Y.; Jiang, C.; Trudeau, S.J.; Narita, Y.; Zhao, B.; Teng, M.; Guo, R.; Gewurz, B.E. Histone Loaders CAF1 and HIRA Restrict Epstein-Barr Virus B-Cell Lytic Reactivation. mBio 2020, 11, e01063-20.
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481.
- De Gorter, D.J.; Vos, J.C.; Pals, S.T.; Spaargaren, M. The B cell antigen receptor controls AP-1 and NFAT activity through Ras-mediated activation of Ral. J. Immunol. 2007, 178, 1405–1414.
- Chen, Y.; Fachko, D.; Ivanov, N.S.; Skinner, C.M.; Skalsky, R.L. Epstein-Barr virus microRNAs regulate B cell receptor signal transduction and lytic reactivation. PLoS Pathog. 2019, 15, e1007535.
More
Information
Subjects:
Virology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Revisions:
2 times
(View History)
Update Date:
07 Oct 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No