 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Massimo Genovese | + 2491 word(s) | 2491 | 2021-08-18 06:04:44 | | | |
| 2 | Dean Liu | Meta information modification | 2491 | 2021-09-29 12:01:53 | | |
Video Upload Options
The tyrosine phosphatase 1B (PTP1B) acts as a key negative regulator of insulin receptor, and a plethora of studies confirmed that uncontrolled activity of this enzyme is one of the main causes that lead to IR (insulin resistance). According to this hypothesis, it has been demonstrated that the overexpression of PTP1B promotes IR in liver, muscle, adipose tissue, pancreas, and brain.
1. Introduction
Type 2 diabetes is a complex pathology characterized by hyperglycemia and metabolic abnormalities affecting different organs and tissues, such as liver, muscle, adipose tissue, and pancreas. To date, subjects affected by T2D can rely on several oral antihyperglycemic drugs showing different mechanisms of action to keep glycaemia under control. These drugs include: inhibitors of intestinal α-glucosidases, which delay intestinal absorption of glucose; metformin, which blocks hepatic gluconeogenesis; different types of secretagogues that stimulate the release of insulin from pancreatic β-cells; thiazolidinediones, which stimulate the storage of circulating fatty acids into adipocytes, thereby improving insulin sensitivity in several peripheral tissues; and the sodium/glucose cotransporter 2 (SGLT-2) inhibitors, which impair the re-uptake of glucose in the renal tubules [1]. The choice of the most appropriate hypoglycemic drug for a patient depends on several factors, such as the patient’s general condition, the presence of comorbidities, tolerance, and the patient’s response to the drug. Generally, most diabetic patients showing hyperglycemia without further pathological complications respond positively to single-drug based therapy ( Figure 1 ), experiencing a decrease of blood sugar levels and an improvement of their general conditions.
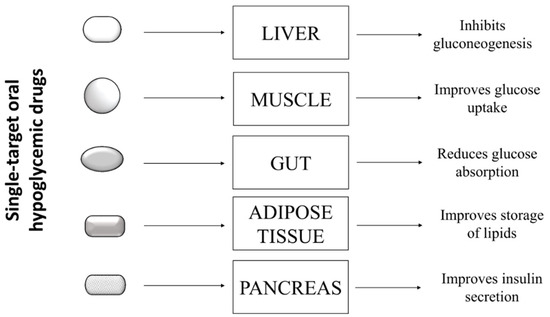
However, many clinical studies showed that benefits obtained with this approach are transient, and in the medium to long term, patients experience a gradual rise in blood sugar and a worsening of general health conditions. In some cases, the up-scaling of drug dosage can allow regaining of the glycemic target, with the hope that, at the same time, no adverse effects related to high doses of the drug occur [2]. The failure of mono-drug therapy is mainly due to the inability of such drugs to replace physiological functions of insulin. Indeed, even if these drugs are able to compensate a specific metabolic defect, they unexpectedly induce severe unbalance in other metabolic pathways.
Very recently, Qi Pan and coworkers evaluated the antidiabetic activity of GLP-1-Fc-FGF21 on diabetic and obese mice models. This new dual targeting agonist, able to target both the GLP-1 and FGF21 ( Fibroblast growth factor 2) pathway, showed a potent antihyperglycemic activity and caused a marked weight loss, suppressing the appetite and reducing caloric intake. Together, these results suggested that GLP-1/FGF21 dual agonists possess all characteristics to become promising new drugs to fight diabetes and obesity [3].
Monosaccharides, such as glucose, fructose, and galactose, are the only sugars absorbed by gut. Oligosaccharides derived from starch digestion are processed by pancreatic α-amylase and intestinal α-glucosidase to produce free glucose that is then uploaded from intestinal cells. Therefore, the rate of blood glucose raising mainly depends on the gut glucose concentration that, in turn, is influenced by the activity of glucosidases present in the gut. This finding inspired many researchers to challenge glucosidase inhibitors as pharmaceutical tools for the treatment of T2D based on the hypothesis that such molecules could delay the release of glucose from complex carbohydrates, slowing down the rise in blood sugar levels observed after a meal. In the last decades, different kinds of glucosidases inhibitors have been produced and approved as antihyperglycemic drugs [4]. Today, such molecules are used as first-line therapy for T2D patients or administrated in combination with other oral anti-diabetics drugs when metformin/biguanides mono-drug based therapies failed the achievement of the glycemic goal [5]. The evidence that glucosidases inhibitors act synergistically with different oral antihyperglycemic drugs suggested that α-glucosidase/PTP1B dual inhibitors could be successfully projected and used as drugs for treatment of T2D.
2. Nature-Inspired Scaffold Molecules for the Synthesis of Dual α-Glucosidase/PTP1B Inhibitors
The role of the OH groups in determining the inhibitory activity of lignanamides is evidenced for compounds showing a phenyldihydronaphthalene core, such as Limoniumin H ( 39), I ( 40), Cannabisin D ( 29), B ( 27), and C ( 28). Cannabisin B, bearing two OH groups on C3 and C3′, showed a high affinity for both targets (IC 50 values for PTP1B and α-glucosidase were 5.89 and 4.56 µM, respectively), whereas compounds ( 28) and ( 39), bearing respectively a methoxyl group on C3′ and on C3, exhibited higher IC 50 values for α-glucosidase. Moreover, the introduction of two methoxyl groups (on C3 and C3′) cause a strong loss of inhibitory activity of compound ( 29). It is interesting to note that the affinity of compound ( 40) for α-glucosidase is eight times lower than ( 39), suggesting that the position of OH groups on naphthalene moiety can influence the interaction of lignanamides with this enzyme.
As the PTP1B inhibitory activity is concerned, all dicaffeoylquinic acids reported in the Table 1 showed significant inhibitory activity. Among these, 1,5 dicaffeoylquinic acid ( 215), the only molecule of this series bearing a caffeic acid molecule linked to C1 of quinic acid, showed the lowest activity overall. Taking into account the differences between the IC 50 values and chemical structure of compounds reported in Table 1, it is reasonable to hypothesize that the presence of the caffeoyl group at the positions 3, 4, or 5 are related to the higher inhibitory activity of 3,4 and 3,5 caffeoyl derivatives for PTP1B.
| Compound | IC50 (µM) | Reference | ||
|---|---|---|---|---|
| PTP1B | α-Glucosidase | |||
| 215 | 1,5-Dicaffeoylquinic acid | 16.05 ± 1.45 | 146.06 ± 0.07 | [6] |
| 216 | 3,4-Dicaffeoylquinic acid | 2.60 ± 0.24 | 128.07 ± 1.67 | [6] |
| 217 | 3,5-Dicaffeoylquinic acid | 2.02 ± 0.46 | 217.40 ± 5.45 | [6] |
| 218 | 4,5-Dicaffeoylquinic acid | 3.21 ± 0.23 | 229.94 ± 1.32 | [6] |
| 219 | Methyl-3,5-di-O-caffeoylquinic acid | 2.99 ± 0.42 | 86.95 ± 4.10 | [6] |
Although dicaffeoylquinic acids ( 215– 219) were also active on α-glucosidase, their inhibitory activity is weaker when compared with that observed on PTP1B. The most potent α-glucosidase inhibitor identified between this group of compounds was the methyl-3,5-di- O -caffeoylquinic acid ( 219). Considering the structural differences of ( 219) with 3,5-dicaffeoylquinic acid ( 217), it is possible to infer that the methyl ester bridge at the carboxylic acid is functional to enhance the inhibitory activity against α-glucosidase.
It is interestingly to observe that among all carbazole, Clausenanisine A ( 224) exhibited the lowest IC 50 values on both targets, suggesting that the carbazole moiety bearing a five-membered cyclic ether, a methoxy group, and a short aliphatic chain represents a promising lead structure for developing new MLDs active on both PTP1B and α-glucosidase. Taking into account that Clausenanisine B ( 225) showed inhibitory activity similar to Clausenanisine A ( 224), we argue that the insertion of a tetrahydro-pyran-4-one group resulted in a slight decrease of the affinity for both targets. The loss of the OH group from C2′ of Clausenanisine B ( 225), of both OH and carbonyl groups, or the introduction of a methoxyl or hydroxyl group on C8 of carbazole moiety generated Euchrestifoline ( 236), Dihydromupamine ( 235), Clauraila B ( 221), and Kurryame ( 237), whose affinity for PTP1B and α-glucosidase steadily decreased. However, the most relevant decrease of affinity occurred after the loss of the carbonic group present on the pyrene moiety, suggested that ketocarbonyl group on C1′ of Clausenanisine B is responsible of the significant inhibitory activity of molecule for both enzymes. Clausenanisine F ( 229), which bears a carboxyl group on C3 and an OH group on C1, showed lower but always significant inhibitory activity for PTP1B while it proved to be a very weak inhibitor of α-glucosidase. The addition of a methoxy group on C2 or C6 of ( 229) leads to Clausenanisine C ( 226) and Clausenaline E ( 222), two molecules with different inhibitory activity. The first one showed a weak affinity for PTP1B but behaved as a potent α-glucosidase inhibitor. The latter showed a reduced affinity for both PTP1B α-glucosidase when compared to ( 229). Finally, Clausenaline F ( 223), bearing two methoxy groups on C1 and C2, and Clausines B ( 232), possessing 2 methoxy groups on C6 and C8, showed a decreased affinity for PTP1B in comparison with ( 229). However, Clausines B resulted a better α-glucosidase inhibitor than Clausenaline F. The replacement of the carboxyl group of ( 229) with a formyl group leads to 3-Formyl-1-hydroxycarbazole ( 220), which showed an affinity for PTP1B similar to ( 229) but an increased affinity for α-glucosidase. The loss of the OH on C1 of ( 231) leads to Clausenanisine D ( 227), which showed a higher IC 50 value for PTP1B but a similar affinity for the α-glucosidase compared to ( 220). Changing the position of one or both OH groups present on ( 220) we obtain Clauszoline N ( 234) and Clauszoline M ( 233). The affinity of these compounds is similar, but that for α-glucosidase differs, ( 233) being a very bad inhibitor of this enzyme. The addition of one methoxy group on C6 of 3-Formyl-1-hydroxycarbazole ( 220) leads to Clausine I ( 230), which possessed a similar affinity for PTP1B as ( 220) but an enhanced affinity for α-glucosidase compared to 3-Formyl-1-hydroxycarbazole. Finally, the introduction of two methoxy groups on C6 and C8 of Clauszoline M ( 233) leads to Clausines B ( 232), a molecule that showed an affinity for PTP1B similar to that of ( 233) but a high affinity for α-glucosidase.
3. Mechanism of Action and In-Vivo Activity of Some Natural PTP1B/α-Glucosidase Inhibitors
Kinetic analyses revealed that these compounds acted as non-competitive inhibitors of PTP1B, while only Phlorofucofuroeckol-A and 7-Phloroeckol behaved as non-competitive inhibitors of α-glucosidase. Conversely, Dieckol acted as competitive inhibitor of the latter. A recent investigation on rat insulinoma cells showed that treatment with Dieckol reduced oxidative stress and apoptosis caused by exposition of cells to high glucose levels, suggesting that this compound could protect β-pancreatic cells against damages induced by hyperglycemia [7].
A recent in-vivo study demonstrated that Phlorofucofuroeckol-A delayed intestinal absorption of dietary carbohydrates in diabetic mice models, suggesting that this molecule could be used to generate new drugs able to reduce post-prandial glucose levels in diabetic patients [8]. High blood glucose levels increase the production of advanced glycation end products that, in turn, contribute to the onset of numerous diabetes-related complications, such as nephropathy, retinopathy, atherosclerosis, and neurodegenerative diseases. Recently, Su Hui Seong et al. demonstrated that Phlorofucofuroeckol-A inhibits non-enzymatic insulin glycation and Aβ aggregation. This finding suggests that this molecule could be used to preserve insulin function and prevent aggregation of the Aβ peptide, thus maintaining the vitality of neurons and avoiding Aβ-mediated brain damage [9]. Besides, it has been demonstrated that Phlorofucofuroeckol-A acts as a potent non-competitive inhibitor of human monoamine oxidase-A, confirming that this compound could be useful to prevent neuronal disorders [10]. A further study showed that Phlorofucofuroeckol-A is effective in contrasting negative effects induced by high fatty diet as well as in reducing leptin resistance in hypothalamic neurons and microglia. This evidence suggests that such compound could be used to fight leptin resistance and to control weight in obese subjects [11].
Manh Tuan Ha and co-workers demonstrated that these compounds act as non-competitive inhibitors of PTP1B and as competitive inhibitors of α-glucosidase [12]. Besides, in-silico docking analyses revealed that all three compounds are docked into the allosteric binding site previously described by Wiesmann et al. in 2004 [13]. Although no data have been produced to confirm the in-vivo activity of such compounds, such results could inspire the synthesis of new MDLs for treatment of type 2 diabetes [12].
Ugonin J ( 190) together with other similar derivatives are flavonoids with cyclohexyl motif extracted from the rhizome part of Helminthostachys zeylanicawas . Abdul Bari Shah and co-workers demonstrated that this compound acts as a competitive inhibitor of PTP1B and non-competitive inhibitor of α-glucosidase [14].
4. Conclusions
MDLs are considered promising alternatives to traditional drugs for the treatment of multifactorial disease. However, the production of new MDLs is not a simple matter. The main obstacle that researchers face in the initial phase of design and synthesis of new drugs is the selection of appropriate scaffold molecules that exhibit at least one initial activity against the selected targets. Sometimes this phase can be very expensive and time consuming, two factors that can lead to the failure of the entire project. In this contest, there is a large consensus in considering the natural world an important source of scaffold molecules to be used for design new MDLs. Data reported in this review demonstrated that many natural molecules possess intrinsic dual α-glucosidase/PTP1B-inhibitory activity. Although most of these possess sub-optimal properties, such as low bioavailability and unfavorable pharmacokinetic or a lowspecificity, semi- or fully synthetic superior derivatives could be easily obtained starting from them. This approach could allow researchers to quickly overcome the first experimental phase, allowing them to focus on the optimization phase aimed at balancing the activity of the molecules toward the biological targets, increasing theirspecificityor bioavailability.
The data we reported confirmed that many natural molecules, including some lignanamides, xanthones, anthraquinones, and several phenolic compounds, show high/balanced inhibitory activity toward PTP1B and α-glucosidase, making these as interesting lead compounds for synthesis of new MDLs. Most of these compounds are characterized by the presence of several hydroxylated phenolic rings or aliphatic chains. What often emerges from SAR analyses is that the number and the position of hydroxyl groups or aliphatic chains are essential to ensure a close interaction between the inhibitors and both enzymes and that by changing one of these parameters, it is possible to influence both inhibitory power and specificity for targets. Moreover, we found that complexity and molecular weight are two parameters strictly related with the IC 50 values of molecules, confirming that the higher the number of OH groups, the higher the affinity for targets. However, small molecules such as Alaternin, Ugonin J, or α-Mangostin also showed high affinity for both targets, suggesting that it is possible to reach a high inhibitory activity also starting from small scaffold molecules. This is another interesting aspect concerning the mechanism of action of these molecules. Data reported in Table 2 shows that smaller molecules mainly behave as competitive inhibitors for PTP1B and bigger ones as non-competitive inhibitors. This evidence suggests that small molecules bearing a hydroxylated phenyl ring could mimic the phenolic structure of the natural substrate, the phosphotyrosine, and for this reason, they could interact more easily with the active site of the enzyme. Conversely, larger molecules, due to their steric hindrance, would not be able to access the active site but could bind to allosteric sites on the surface of the enzyme.
| Compound | Inhibition Type | |
|---|---|---|
| PTP1B | α-Glucosidase | |
| Alaternin | Competitive | Mixed-type inhibition |
| Albanol B | Mixed-type inhibition | Mixed-type inhibition |
| Albasin B | Non competitive | Competitive |
| Dieckol | Non competitive | Competitive |
| Murasalbin D | Non competitive | Competitive |
| 7-Phloroeckol | Non competitive | Non competitive |
| Macrourin G | Non competitive | Competitive |
| 6-geranyl-3,3′,5,5′,7-pentahydroxy-4′-methoxyflavane | Mixed-type inhibition | Non competitive |
| Phlorofucofuroeckol-A | Non competitive | Non competitive |
| Ugonin J | Competitive | Non competitive |
| Ugonin M | Mixed-type inhibition | Mixed-type inhibition |
To the best of our knowledge, few synthetic PTP1B/α-glucosidase inhibitors have been produced still to date, and none of these has been evaluated in clinical trials to date, which is probably due to the fact that development of such kinds of molecules is still in its infancy. Researchers hope that the information reported in this entry will be useful to researchers dedicated to the design and synthesis of novel dual PTP1B/α-glucosidase inhibitors to be used routinely for the treatment of patients affected by T2D, obesity, and metabolic syndrome.
References
- Brunetti, L.; Kalabalik, J. Management of type-2 diabetes mellitus in adults: Focus on individualizing non-insulin therapies. Pharm. Ther. 2012, 37, 687–696.
- Del Prato, S.; Felton, A.-M.; Munro, N.; Nesto, R.; Zimmet, P.; Zinman, B.; Global Partnership for Effective Diabetes Management. Improving glucose management: Ten steps to get more patients with type 2 diabetes to glycaemic goal. Int. J. Clin. Pract. 2005, 59, 1345–1355.
- Pan, Q.; Lin, S.; Li, Y.; Liu, L.; Li, X.; Gao, X.; Yan, J.; Gu, B.; Chen, X.; Li, W.; et al. A novel GLP-1 and FGF21 dual agonist has therapeutic potential for diabetes and non-alcoholic steatohepatitis. EBioMedicine 2021, 63, 103202.
- Joshi, S.R.; Standl, E.; Tong, N.; Shah, P.; Kalra, S.; Rathod, R. Therapeutic potential of α-glucosidase inhibitors in type 2 diabetes mellitus: An evidence-based review. Expert Opin. Pharmacother. 2015, 16, 1959–1981.
- Yang, H.K.; Lee, S.H.; Shin, J.; Choi, Y.H.; Ahn, Y.B.; Lee, B.W.; Rhee, E.J.; Min, K.W.; Yoon, K.H. Acarbose Add-on Therapy in Patients with Type 2 Diabetes Mellitus with Metformin and Sitagliptin Failure: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Study. Diabetes Metab. J. 2019, 43, 287–301.
- Nurul Islam, M.; Jung, H.A.; Sohn, H.S.; Kim, H.M.; Choi, J.S. Potent α-glucosidase and protein tyrosine phosphatase 1B inhibitors from Artemisia capillaris. Arch. Pharm. Res. 2013, 36, 542–552.
- Lee, S.-H.; Park, M.-H.; Kang, S.-M.; Ko, S.-C.; Kang, M.-C.; Cho, S.; Park, P.-J.; Jeon, B.-T.; Kim, S.-K.; Han, J.-S.; et al. Dieckol isolated from Ecklonia cava protects against high-glucose induced damage to rat insulinoma cells by reducing oxidative stress and apoptosis. Biosci. Biotechnol. Biochem. 2012, 76, 1445–1451.
- You, H.-N.; Lee, H.-A.; Park, M.-H.; Lee, J.-H.; Han, J.-S. Phlorofucofuroeckol A isolated from Ecklonia cava alleviates postprandial hyperglycemia in diabetic mice. Eur. J. Pharmacol. 2015, 752, 92–96.
- Seong, S.H.; Paudel, P.; Jung, H.A.; Choi, J.S. Identifying Phlorofucofuroeckol-A as a Dual Inhibitor of Amyloid-β25-35 Self-Aggregation and Insulin Glycation: Elucidation of the Molecular Mechanism of Action. Mar. Drugs 2019, 17, 600.
- Seong, S.H.; Paudel, P.; Choi, J.-W.; Ahn, D.H.; Nam, T.-J.; Jung, H.A.; Choi, J.S. Probing Multi-Target Action of Phlorotannins as New Monoamine Oxidase Inhibitors and Dopaminergic Receptor Modulators with the Potential for Treatment of Neuronal Disorders. Mar. Drugs 2019, 17, 377.
- Oh, S.; Son, M.; Choi, J.; Choi, C.H.; Park, K.Y.; Son, K.H.; Byun, K. Phlorotannins from Ecklonia cava Attenuates Palmitate-Induced Endoplasmic Reticulum Stress and Leptin Resistance in Hypothalamic Neurons. Mar. Drugs 2019, 17, 570.
- Ha, M.T.; Seong, S.H.; Nguyen, T.D.; Cho, W.-K.; Ah, K.J.; Ma, J.Y.; Woo, M.H.; Choi, J.S.; Min, B.S. Chalcone derivatives from the root bark of Morus alba L. act as inhibitors of PTP1B and α-glucosidase. Phytochemistry 2018, 155, 114–125.
- Wiesmann, C.; Barr, K.J.; Kung, J.; Zhu, J.; Erlanson, D.A.; Shen, W.; Fahr, B.J.; Zhong, M.; Taylor, L.; Randal, M.; et al. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730–737.
- Shah, A.B.; Yoon, S.; Kim, J.H.; Zhumanova, K.; Ban, Y.J.; Lee, K.W.; Park, K.H. Effectiveness of cyclohexyl functionality in ugonins from Helminthostachys zeylanica to PTP1B and α-glucosidase inhibitions. Int. J. Biol. Macromol. 2020, 165, 1822–1831.

