 +1 credit
+1 credit
Video Upload Options
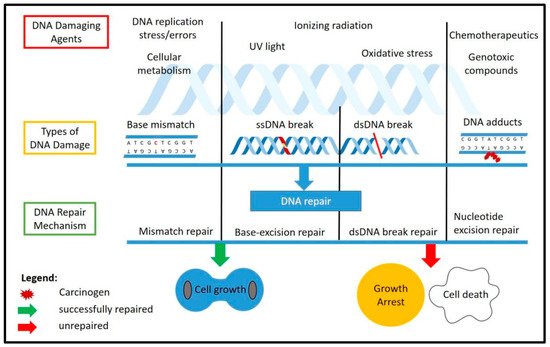
DNA damage could occur in cells either endogenously, through normal cellular replication and metabolism, or exogenously through ultraviolet (UV), ionizing radiation (IR) or various genotoxic compounds] that could induce DNA damage. Different stressors will cause different types of DNA damage. Normal DNA replication could induce mismatch of the nucleotide and cause mutations. Stressors such as oxidative stress will produce reactive oxygen species (ROS) from normal cellular metabolism or from external genotoxic compound, which will cause DNA breaks, either single-stranded or double-stranded. Unrepaired DNA damage could cause severe mutations and chromosomal instability, which would have detrimental effects on the cells and lead to cell death, while DNA breaks that are repaired through non-homologous end joining (NHEJ) might cause mutations during the process.
The DDR is the response mechanism which will detect any DNA damage that occurs throughout the chromosome and will activate a repair cascade to the damage site. This will help the cells either to proliferate normally if the repair was successful or to activate the cellular programmed cell death if the damage was too extensive and was unable to be repaired. The known DNA damage repair mechanisms include mismatch repair (MMR), base excision repair (BER), nucleotide excision repair (NER), homologous recombination (HR) and non-homologous end joining (NHEJ). Specific types of DNA damage could be fixed by a specific repair factor, such as the ATM kinase, which is the main factor in double-strand break repair through NHEJ. Figure 1 shows the causes and types of DNA damage as well as the response cascade involved in repairing the damages.
1. DNA Damage Response (DDR)
2. TRF2 Post-Translational Modification and Its Involvement with DDR
| Modification | Amino Acids | Modifying Enzymes | Function | Reference (s) |
|---|---|---|---|---|
| Phosphorylation | Ser20 | Chk2 | Phosphorylation of TRF2 by Chk2 decreases TRF2 binding to telomeric DNA. S20 phosphorylation is required for TRF2 interaction with G-rich RNA and recruitment of ORC to OriP. | [58][59] |
| Ser365 | CDK | Regulates the assembly and disassembly of t-loop during the cell-cycle or DNA replication, which protects the telomeres from replicative stress. | [53] | |
| Thr188 | ATM | Early recruitment at the DNA break site and may be involved in DNA damage response/repair. | [56][82] | |
| Thr358 | Aurora C | Possible function in cell replication and unwinding of the chromosome. | [83] | |
| SUMOylation | IK140TE, LK245SE, and RK333DE | MMS21 | Stabilization of TRF2 and recruitment at the telomeres. | [64][65] |
| - | PIAS1 | Interacts with RNF4 to regulate TRF2 level at the telomeres. | [62] | |
| Ubiquitylation | K173 K180 K184 | Siah1 | Siah1 is a p53-inducible E3 ligase with a C3H4-type RING finger domain. Knockdown of Siah1 stabilizes TRF2 and telomere length maintenance. | [70] |
| - | RNF4 | Interacts together with PIAS1 in modulating TRF2 role in telomere maintenance and protection through regulating the level of TRF2 at the telomeres. Acts downstream of the ATM-kinase pathway in DDR. | [62][69] | |
| Acetylation | K293 | P300 | Required for TRF2 stabilization and efficient binding at the telomeres. | [71] |
| Poly(ADP-ribosyl)ation or PARsylation | - | PARP1, PARP2 | Reduces the binding of TRF2 to telomeres that may contribute to dysfunctional telomeres. | [77][84] |
3. TRF2 Protects Telomeres by Blocking the Activation of DDR
References
- Rothkamm, K.; Krüger, I.; Thompson, L.H.; Löbrich, M. Pathways of DNA Double-Strand Break Repair during the Mammalian Cell Cycle. Mol. Cell. Biol. 2003, 23, 5706–5715.
- Fagagna, F.d.A.d.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198.
- Shimizu, I.; Yoshida, Y.; Suda, M.; Minamino, T. DNA Damage Response and Metabolic Disease. Cell Metab. 2014, 20, 967–977.
- Turgeon, M.-O.; Perry, N.J.S.; Poulogiannis, G. DNA Damage, Repair, and Cancer Metabolism. Front. Oncol. 2018, 8, 15.
- Sinha, R.P.; Häder, D.-P. UV-induced DNA damage and repair: A review. Photochem. Photobiol. Sci. 2002, 1, 225–236.
- Rastogi, R.P.; Richa; Kumar, A.; Tyagi, M.B.; Sinha, R.P. Molecular Mechanisms of Ultraviolet Radiation-Induced DNA Damage and Repair. J. Nucleic Acids 2010, 2010, 1–32.
- Sachs, R.K.; Chen, P.-L.; Hahnfeldt, P.J.; Hlatky, L.R. DNA damage caused by ionizing radiation. Math. Biosci. 1992, 112, 271–303.
- Leadon, S.A. Repair of DNA damage produced by ionizing radiation: A minireview. Semin. Radiat. Oncol. 1996, 6, 295–305.
- Awang, N.; Kismin, D.N.A.; Kamaludin, N.F.; Ghazali, A.R.b. Genotoxic Effects on buccal Cells of Workers Exposed to Fogging Sprays during Fogging Operation. Biomed. J. Sci. Tech. Res. 2017, 1, 1341–1345.
- Sopian, N.; Jalaludin, J.; Abu Bakar, S.; Hamedon, T.; Latif, M. Exposure to Particulate PAHs on Potential Genotoxicity and Cancer Risk among School Children Living Near the Petrochemical Industry. Int. J. Environ. Res. Public Health 2021, 18, 2575.
- Morrison, A.; Johnson, A.; Johnston, L.; Sugino, A. Pathway correcting DNA replication errors in Saccharomyces cerevisiae. EMBO J. 1993, 12, 1467–1473.
- Brown, T.A. Mutation, Repair and Recombination. In Genomes, 2nd ed.; Wiley-Liss: Oxford, UK, 2002.
- Boyce Kylie, J.; Wang, Y.; Verma, S.; Shakya Viplendra, P.S.; Xue, C.; Idnurm, A.; Alspaugh, J.A. Mismatch Repair of DNA Replication Errors Contributes to Microevolution in the Pathogenic Fungus Cryptococcus neoformans. mBio 2017, 8, e00595-17.
- Kunkel, T.A.; Erie, D.A. Eukaryotic Mismatch Repair in Relation to DNA Replication. Annu. Rev. Genet. 2015, 49, 291–313.
- Yang, J.; Yu, Y.; Hamrick, H.E.; Duerksen-Hughes, P.J. ATM, ATR and DNA-PK: Initiators of the cellular genotoxic stress responses. Carcinogenesis 2003, 24, 1571–1580.
- Coates, P.J.; Lorimore, S.A.; Wright, E.G. Cell and tissue responses to genotoxic stress. J. Pathol. 2005, 205, 221–235.
- Tsolou, A.; Nelson, G.; Trachana, V.; Chondrogianni, N.; Saretzki, G.; von Zglinicki, T.; Gonos, E.S. The 19S proteasome subunit Rpn7 stabilizes DNA damage foci upon genotoxic insult. IUBMB Life 2012, 64, 432–442.
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631.
- Anindya, R. Single-stranded DNA damage: Protecting the single-stranded DNA from chemical attack. DNA Repair 2020, 87, 102804.
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714.
- Li, G.-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98.
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harbor Perspect. Biol. 2013, 5, a012583.
- Spivak, G. Nucleotide excision repair in humans. DNA Repair 2015, 36, 13–18.
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535.
- Chiruvella, K.K.; Liang, Z.; Wilson, T.E. Repair of Double-Strand Breaks by End Joining. Cold Spring Harb. Perspect. Biol. 2013, 5, a012757.
- Rondeau, S.; Vacher, S.; De Koning, L.; Briaux, A.; Schnitzler, A.; Chemlali, W.; Callens, C.; Lidereau, R.; Bièche, I. ATM has a major role in the double-strand break repair pathway dysregulation in sporadic breast carcinomas and is an independent prognostic marker at both mRNA and protein levels. Br. J. Cancer 2015, 112, 1059–1066.
- Niida, H.; Nakanishi, M. DNA damage checkpoints in mammals. Mutagenesis 2005, 21, 3–9.
- Nastasi, C.; Mannarino, L.; D’Incalci, M. DNA Damage Response and Immune Defense. Int. J. Mol. Sci. 2020, 21, 7504.
- Andrs, M.; Korabecny, J.; Jun, D.; Hodny, Z.; Bartek, J.; Kuca, K. Phosphatidylinositol 3-Kinase (PI3K) and Phosphatidylinositol 3-Kinase-Related Kinase (PIKK) Inhibitors: Importance of the Morpholine Ring. J. Med. Chem. 2015, 58, 41–71.
- Lovejoy, C.A.; Cortez, D. Common mechanisms of PIKK regulation. DNA Repair 2009, 8, 1004–1008.
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs kinases—The lessons from the mouse models: Inhibition ≠ deletion. Cell Biosci. 2020, 10, 1–15.
- Guo, F.; Li, J.; Du, W.; Zhang, S.; O’Connor, M.; Thomas, G.; Kozma, S.; Zingarelli, B.; Pang, Q.; Zheng, Y. mTOR regulates DNA damage response through NF-κB-mediated FANCD2 pathway in hematopoietic cells. Leukemia 2013, 27, 2040–2046.
- Alao, J.-P.; Legon, L.; Rallis, C. Crosstalk between the mTOR and DNA Damage Response Pathways in Fission Yeast. Cells 2021, 10, 305.
- Hiom, K. DNA Repair: How to PIKK a Partner. Curr. Biol. 2005, 15, R473–R475.
- Klapper, W.; Qian, W.; Schulte, C.; Parwaresch, R. DNA damage transiently increases TRF2 mRNA expression and telomerase activity. Leukemia 2003, 17, 2007–2015.
- Saha, B.; Zitnik, G.; Johnson, S.; Nguyen, Q.; Risques, R.A.; Martin, G.M.; Oshima, J. DNA damage accumulation and TRF2 degradation in atypical Werner syndrome fibroblasts with LMNA mutations. Front. Genet. 2013, 4, 129.
- Takai, H.; Smogorzewska, A.; de Lange, T. DNA Damage Foci at Dysfunctional Telomeres. Curr. Biol. 2003, 13, 1549–1556.
- Dinami, R.; Porru, M.; Amoreo, C.A.; Sperduti, I.; Mottolese, M.; Buglioni, S.; Marinelli, D.; Maugeri-Saccà, M.; Sacconi, A.; Blandino, G.; et al. TRF2 and VEGF-A: An unknown relationship with prognostic impact on survival of colorectal cancer patients. J. Exp. Clin. Cancer Res. 2020, 39, 1–13.
- Diehl, M.C.; Idowu, M.O.; Kimmelshue, K.N.; York, T.P.; Jackson-Cook, C.K.; Turner, K.C.; Holt, S.E.; Elmore, L.W. Elevated TRF2 in advanced breast cancers with short telomeres. Breast Cancer Res. Treat. 2010, 127, 623–630.
- Nijjar, T.; Bassett, E.; Garbe, J.; Takenaka, Y.; Stampfer, M.R.; Gilley, D.; Yaswen, P. Accumulation and altered localization of telomere-associated protein TRF2 in immortally transformed and tumor-derived human breast cells. Oncogene 2005, 24, 3369–3376.
- Knecht, H.; Sawan, B.; Lichtensztejn, D.; Lemieux, B.; Wellinger, R.J.; Mai, S. The 3D nuclear organization of telomeres marks the transition from Hodgkin to Reed–Sternberg cells. Leukemia 2008, 23, 565–573.
- Lajoie, V.; Lemieux, B.; Sawan, B.; Lichtensztejn, D.; Lichtensztejn, Z.; Wellinger, R.; Mai, S.; Knecht, H. LMP1 mediates multinuclearity through downregulation of shelterin proteins and formation of telomeric aggregates. Blood 2015, 125, 2101–2110.
- Knecht, H.; Johnson, N.A.; Haliotis, T.; Lichtensztejn, D.; Mai, S. Disruption of direct 3D telomere–TRF2 interaction through two molecularly disparate mechanisms is a hallmark of primary Hodgkin and Reed–Sternberg cells. Lab. Investig. 2017, 97, 772–781.
- Muñoz, P.; Blanco, R.; Flores, J.M.; Blasco, M.A. XPF nuclease-dependent telomere loss and increased DNA damage in mice overexpressing TRF2 result in premature aging and cancer. Nat. Genet. 2005, 37, 1063–1071.
- Wang, Z.; Wu, X. Abnormal function of telomere protein TRF2 induces cell mutation and the effects of environmental tumor-promoting factors (Review). Oncol. Rep. 2021, 46, 1–20.
- Matsutani, N.; Yokozaki, H.; Tahara, E.; Tahara, H.; Kuniyasu, H.; Haruma, K.; Chayama, K.; Yasui, W.; Tahara, E. Expression of telomeric repeat binding factor 1 and 2 and TRF1-interacting nuclear protein 2 in human gastric carcinomas. Int. J. Oncol. 2001, 19, 507–512.
- Bellon, M.; Datta, A.; Brown, M.; Pouliquen, J.-F.; Couppié, P.; Kazanji, M.; Nicot, C. Increased expression of telomere length regulating factors TRF1, TRF2 and TIN2 in patients with adult T-cell leukemia. Int. J. Cancer 2006, 119, 2090–2097.
- Nera, B.; Huang, H.-S.; Lai, T.; Xu, L. Elevated levels of TRF2 induce telomeric ultrafine anaphase bridges and rapid telomere deletions. Nat. Commun. 2015, 6, 10132.
- Muhammad Imran, S.A. The Role of TRF2 in Regulating Neural Progenitor Cells Proliferation and Surivival; Imperial College London: London, UK, 2019.
- Walker, J.R.; Zhu, X.-D. Post-translational modifications of TRF1 and TRF2 and their roles in telomere maintenance. Mech. Ageing Dev. 2012, 133, 421–434.
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520.
- Chi, Y.; Welcker, M.; Hizli, A.A.; Posakony, J.J.; Aebersold, R.; Clurman, B.E. Identification of CDK2 substrates in human cell lysates. Genome Biol. 2008, 9, R149.
- Sarek, G.; Kotsantis, P.; Ruis, P.; Van Ly, D.; Margalef, P.; Borel, V.; Zheng, X.-F.; Flynn, H.; Snijders, B.; Chowdhury, D.; et al. CDK phosphorylation of TRF2 controls t-loop dynamics during the cell cycle. Nat. Cell Biol. 2019, 575, 523–527.
- Picco, V.; Coste, I.; Giraud-Panis, M.-J.; Renno, T.; Gilson, E.; Pagès, G. ERK1/2/MAPK pathway-dependent regulation of the telomeric factor TRF. Oncotarget 2016, 7, 46615–46627.
- Kim, S.-T.; Lim, D.-S.; Canman, C.E.; Kastan, M.B. Substrate Specificities and Identification of Putative Substrates of ATM Kinase Family Members. J. Biol. Chem. 1999, 274, 37538–37543.
- Tanaka, H.; Mendonca, M.S.; Bradshaw, P.S.; Hoelz, D.J.; Malkas, L.H.; Meyn, M.S.; Gilley, D. DNA damage-induced phosphorylation of the human telomere-associated protein TRF. Proc. Natl. Acad. Sci. USA 2005, 102, 15539–15544.
- Matsuoka, S.; Rotman, G.; Ogawa, A.; Shiloh, Y.; Tamai, K.; Elledge, S.J. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 10389–10394.
- Buscemi, G.; Zannini, L.; Fontanella, E.; Lecis, D.; Lisanti, S.; Delia, D. The Shelterin Protein TRF2 Inhibits Chk2 Activity at Telomeres in the Absence of DNA Damage. Curr. Biol. 2009, 19, 874–879.
- Zhou, J.; Deng, Z.; Norseen, J.; Lieberman, P.M. Regulation of Epstein-Barr Virus Origin of Plasmid Replication (OriP) by the S-Phase Checkpoint Kinase Chk. J. Virol. 2010, 84, 4979–4987.
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956.
- Yang, Y.; He, Y.; Wang, X.; Liang, Z.; He, G.; Zhang, P.; Zhu, H.; Xu, N.; Liang, S. Protein SUMOylation modification and its associations with disease. Open Biol. 2017, 7, 170167.
- Her, J.; Jeong, Y.Y.; Chung, I.K. PIAS1-mediated sumoylation promotes STUbL-dependent proteasomal degradation of the human telomeric protein TRFFEBS. Letters 2015, 589, 3277–3286.
- Rai, R.; Chen, Y.; Lei, M.; Chang, S. TRF2-RAP1 is required to protect telomeres from engaging in homologous recombination-mediated deletions and fusions. Nat. Commun. 2016, 7, 10881.
- Potts, P.R.; Yu, H. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat. Struct. Mol. Biol. 2007, 14, 581–590.
- Yalçin, Z.; Selenz, C.; Jacobs, J.J.L. Ubiquitination and SUMOylation in Telomere Maintenance and Dysfunction. Front. Genet. 2017, 8, 67.
- Peuscher, M.; Jacobs, J.J. Posttranslational control of telomere maintenance and the telomere damage response. Cell Cycle 2012, 11, 1524–1534.
- Sun, H.; Leverson, J.D.; Hunter, T. Conserved function of RNF4 family proteins in eukaryotes: Targeting a ubiquitin ligase to SUMOylated proteins. EMBO J. 2007, 26, 4102–4112.
- Chang, Y.-C.; Oram, M.; Bielinsky, A.-K. SUMO-Targeted Ubiquitin Ligases and Their Functions in Maintaining Genome Stability. Int. J. Mol. Sci. 2021, 22, 5391.
- Groocock, L.M.; Nie, M.; Prudden, J.; Moiani, D.; Wang, T.; Cheltsov, A.; Rambo, R.P.; Arvai, A.S.; Hitomi, C.; Tainer, J.A.; et al. RNF 4 interacts with both SUMO and nucleosomes to promote the DNA damage response. EMBO Rep. 2014, 15, 601–608.
- Fujita, K.; Horikawa, I.; Mondal, A.M.; Jenkins, L.M.M.; Appella, E.; Vojtesek, B.; Bourdon, J.-C.; Lane, D.; Harris, C.C. Positive feedback between p53 and TRF2 during telomere-damage signalling and cellular senescence. Nat. Cell Biol. 2010, 12, 1205–1212.
- Her, Y.R.; Chung, I.K. p300-mediated acetylation of TRF2 is required for maintaining functional telomeres. Nucleic Acids Res. 2013, 41, 2267–2283.
- Dutto, I.; Scalera, C.; Prosperi, E. CREBBP and p300 lysine acetyl transferases in the DNA damage response. Cell. Mol. Life Sci. 2018, 75, 1325–1338.
- Hassa, P.O.; Buerki, C.; Lombardi, C.; Imhof, R.; Hottiger, M.O. Transcriptional Coactivation of Nuclear Factor-κB-dependent Gene Expression by p300 Is Regulated by Poly(ADP)-ribose Polymerase-1*. J. Biol. Chem. 2003, 278, 45145–45153.
- Chaudhuri, A.R.; Nussenzweig, A.R.C.A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621.
- Haince, J.-F.; Kozlov, S.; Dawson, V.; Dawson, T.M.; Hendzel, M.; Lavin, M.F.; Poirier, G.G. Ataxia Telangiectasia Mutated (ATM) Signaling Network Is Modulated by a Novel Poly(ADP-ribose)-dependent Pathway in the Early Response to DNA-damaging Agents. J. Biol. Chem. 2007, 282, 16441–16453.
- Sousa, F.; Matuo, R.; Soares, D.G.; Escargueil, A.; Henriques, J.A.; Larsen, A.K.; Saffi, J. PARPs and the DNA damage response. Carcinogenesis 2012, 33, 1433–1440.
- Gomez, M.; Wu, J.; Schreiber, V.; Dunlap, J.; Dantzer, F.; Wang, Y.; Liu, Y. PARP1 Is a TRF2-associated Poly(ADP-Ribose)Polymerase and Protects Eroded Telomeres. Mol. Biol. Cell 2006, 17, 1686–1696.
- Huda, N.; Abe, S.; Gu, L.; Mendonca, M.S.; Mohanty, S.; Gilley, D. Recruitment of TRF2 to laser-induced DNA damage sites. Free. Radic. Biol. Med. 2012, 53, 1192–1197.
- Kong, X.; Cruz, G.M.S.; Trinh, S.L.; Zhu, X.-D.; Berns, M.W.; Yokomori, K. Biphasic recruitment of TRF2 to DNA damage sites promotes non-sister chromatid homologous recombination repair. J. Cell Sci. 2018, 131, 219311.
- Mao, Z.; Seluanov, A.; Jiang, Y.; Gorbunova, V. TRF2 is required for repair of nontelomeric DNA double-strand breaks by homologous recombination. Proc. Natl. Acad. Sci. USA 2007, 104, 13068–13073.
- Zalzman, M.; Meltzer, W.A.; Portney, B.A.; Brown, R.A.; Gupta, A. The Role of Ubiquitination and SUMOylation in Telomere Biology. Curr. Issues Mol. Biol. 2020, 35, 85–98.
- Huda, N.; Tanaka, H.; Mendonca, M.S.; Gilley, D. DNA Damage-Induced Phosphorylation of TRF2 Is Required for the Fast Pathway of DNA Double-Strand Break Repair. Mol. Cell. Biol. 2009, 29, 3597–3604.
- Spengler, D. The Protein Kinase Aurora-C Phosphorylates TRF. Cell Cycle 2007, 6, 2579–2580.
- Dantzer, F.; Giraud-Panis, M.-J.; Jaco, I.; Amé, J.-C.; Schultz, I.; Blasco, M.; Koering, C.-E.; Gilson, E.; Murcia, J.M.-D.; de Murcia, G.; et al. Functional Interaction between Poly(ADP-Ribose) Polymerase 2 (PARP-2) and TRF2: PARP Activity Negatively Regulates TRF. Mol. Cell. Biol. 2004, 24, 1595–1607.
- Baker, A.M.; Fu, Q.; Hayward, W.; Victoria, S.; Pedroso, I.M.; Lindsay, S.M.; Fletcher, T.M. The Telomere Binding Protein TRF2 Induces Chromatin Compaction. PLoS ONE 2011, 6, e19124.
- De Lange, T. How Telomeres Solve the End-Protection Problem. Science 2009, 326, 948.
- Palm, W.; de Lange, T. How Shelterin Protects Mammalian Telomeres. Annu. Rev. Genet. 2008, 42, 301–334.
- Van Steensel, B.; Smogorzewska, A.; de Lange, T. TRF2 Protects Human Telomeres from End-to-End Fusions. Cell 1998, 92, 401–413.
- Saint-Léger, A.; Koelblen, M.; Civitelli, L.; Bah, A.; Djerbi, N.; Giraud-Panis, M.-J.; Londono-Vallejo, A.; Ascenzioni, F.; Gilson, E. The basic N-terminal domain of TRF2 limits recombination endonuclease action at human telomeres. Cell Cycle 2014, 13, 2469–2474.
- Karlseder, J.; Hoke, K.; Mirzoeva, O.K.; Bakkenist, C.; Kastan, M.B.; Petrini, J.; De Lange, T. The Telomeric Protein TRF2 Binds the ATM Kinase and Can Inhibit the ATM-Dependent DNA Damage Response. PLoS Biol. 2004, 2, e240.
- Arnoult, N.; Karlseder, J. Complex interactions between the DNA-damage response and mammalian telomeres. Nat. Struct. Mol. Biol. 2015, 22, 859–866.
- Blasco, M.A. Telomeres and human disease: Ageing, cancer and beyond. Nat. Rev. Genet. 2005, 6, 611–622.
- Celli, G.B.; de Lange, T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 2005, 7, 712–718.
- Denchi, E.L.; de Lange, T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT. Nature 2007, 448, 1068–1071.
- Hewitt, G.; Jurk, D.; Marques, F.M.; Correia-Melo, C.; Hardy, T.L.D.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708.
- Okamoto, K.; Bartocci, C.; Ouzounov, I.; Diedrich, J.K.; Iii, J.R.Y.; Denchi, E.L. A two-step mechanism for TRF2-mediated chromosome-end protection. Nat. Cell Biol. 2013, 494, 502–505.
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, 100492.

