 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dong Zhang | + 5917 word(s) | 5917 | 2020-07-28 10:42:01 | | | |
| 2 | Dong Zhang | + 1 word(s) | 5918 | 2020-07-28 15:04:31 | | | | |
| 3 | Dong Zhang | + 1 word(s) | 5918 | 2020-07-28 15:08:29 | | | | |
| 4 | Dong Zhang | + 2 word(s) | 5919 | 2020-07-28 15:11:58 | | | | |
| 5 | Rita Xu | -2803 word(s) | 3116 | 2020-07-30 10:46:40 | | |
Video Upload Options
BARD1 is a very important BRCA1 binding partner and plays a key role in the development of a variety of tumors. Similar to BRCA1, BARD1 has been implicated in the development of breast and gynecological cancers. In addition, BARD1 also plays a role in the development of non-breast and non-gynecological cancers.
1. Definition
Breast Cancer 1 (BRCA1) gene is a well-characterized tumor suppressor gene, mutations of which are primarily found in women with breast and ovarian cancers. BRCA1-associated RING domain 1 (BARD1) gene has also been identified as an important tumor suppressor gene in breast, ovarian, and uterine cancers. Underscoring the functional significance of the BRCA1 and BARD1 interactions, prevalent mutations in the BRCA1 gene are found in its RING domain, through which it binds the RING domain of BARD1. BARD1-BRCA1 heterodimer plays a crucial role in a variety of DNA damage response (DDR) pathways, including DNA damage checkpoint and homologous recombination (HR). However, many mutations in both BARD1 and BRCA1 also exist in other domains that significantly affect their biological functions. Intriguingly, recent genome-wide studies have identified various single nucleotide polymorphisms (SNPs), genetic alterations, and epigenetic modifications in or near the BARD1 gene that manifested profound effects on tumorigenesis in a variety of non-breast and non-gynecological cancers.
2. Introduction
BARD1 was discovered in 1996 by Baer’s group who sought out to identify proteins that interacted with the N-terminus of BRCA1 through a two-hybrid screening [1]. Of the 16 candidates identified, only BARD1 was able to directly interact with and form a stable complex with BRCA1 in mammalian cells through its RING domain, hence its name, BRCA1-associated RING domain 1 (BARD1) gene. BARD1 gene is localized to 2q35 and encodes a full-length (FL) protein (BARD1-FL) composed of 777 amino acids (a.a.) [1][2][3]. There are many structural similarities between BRCA1 and BARD1 (Figure 1A). The RING domain of BARD1 consists of a.a. 49–100 and is rich in cysteine and histidine, which bind to two zinc ions [1][4]. However, the RING domain of BARD1 is slightly shorter and lacks the central α-helix as well as the third β-pleated sheet when compared to that of BRCA1. It is flanked by two α-helices (a.a. 36–48 and 101–116), which interact with the helices adjacent to the RING domain of BRCA1 to form a heterodimer. Similar to BRCA1, BARD1 also contains two tandem copies of the BRCA1 C-terminal (BRCT) domain located at a.a. 568–777 [5][6]. The domain is composed of BRCT1 (a.a. 568–654) connected to BRCT2 (a.a. 669–777) by an α-helix. BRCT1 has a hydrophilic binding pocket, which binds to phospho-peptides, just like BRCA1. The second binding pocket is located between the two BRCT domains and is relatively hydrophobic. The amino acid sequences of the second binding pocket of BARD1 are different from those in BRCA1, suggesting unique ligand interactions. BARD1, like BRCA1, contains a nuclear export sequence (NES) at a.a. 102–120 that allows for its transport out of the nucleus and into the cytoplasm [7]. It also contains six nuclear localization signals (NLS) which allow the protein to move back into the nucleus [8]. The NLS sequences are located at a.a. 127–130, 139–155, 321–337, 365–371, 657–663, and 706–709.
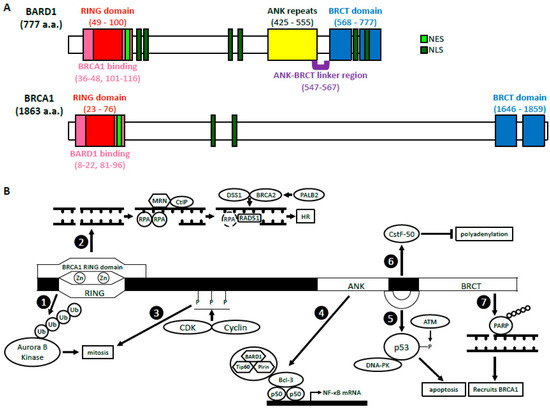
Figure 1. The structure and functions of BARD1 (A) BARD1 and BRCA1 domain structures. BARD1 and BRCA1 have similar RING domains located at their N-termini, BRCT domains located at their C-termini, as well as NES and NLS. The ANK repeats and the ANK-BRCT linker region are unique to BARD1. (B) A summary of the biological functions of BARD1. BRCA1-dependent: The BARD1 RING domain interacts with the RING domain of BRCA1. (1) The E3 ligase activity of BARD1-BRCA1. (2) The HR function of BARD1-BRCA1. BRCA1-independent: (3) CDK2-CyclinA1/E1 and CDK1-CyclinB phosphorylate BARD1, which then facilitates mitotic progression. (4) BARD1 forms a complex with Tip60 and Pirin that interacts with p50 and Bcl-3 through BARD1’s ANK and BRCT domains. This complex binds the NF-κB promoter and regulates its transcription. (5) BARD1, together with DNA-PK, stabilizes p53 through its ANK-BRCT linker region, which allows phosphorylation by ATM and induction of apoptosis. (6) The ANK-BRCT linker region also binds CstF-50 and together they prevent polyadenylation of mRNA in response to DNA damage. (7) BARD1 binds the poly (ADP-ribose) through its BRCT domain and recruits BRCA1 to the DNA damage sites.
BARD1’s structure differs from BRCA1 also due to the presence of ankyrin (ANK) repeats at a.a. 425–555 [6][9]. This domain consists of four ANK repeats that compose a hydrophobic helical core. The fourth ANK repeat differs in its sequence and is shortened but maintains an overall similar structure. The non-traditional orientation of the fourth ANK may be attributed to a span of 20 amino acids (a.a. 547–567), which include and extend beyond the C-terminal ANK repeat domain. This region, which has no secondary structure, is thus called the ANK-BRCT linker region. Overall, the structural domains of BARD1 are critical for their biological functions because they are vital for various protein-protein and protein-DNA interactions.
Due to the pronounced role of BRCA1 and BRCA2 in hereditary breast and ovarian cancer, different mutations and variants of BARD1 were first investigated in breast cancers and various gynecological cancers in the late 1990s and early 2000s [10][11][12]. The genetic changes in BARD1 include missense mutations, nonsense mutations, and deletions. For example, women with the missense mutation C557S, just before the BRCT1 domain of BARD1, have an increased susceptibility to breast cancer [11][12]. Many studies have also identified various isoforms of BARD1 that result from alternative splicing (Figure 2A). The α isoform removes exon 2, while the β isoform splices out exons 2 and 3 [13]. The γ isoform loses either exon 4 [13] or exons 4–11 [14]. The δ isoform lacks exons 2 through 6 [15][16], while the ε isoform removes exons 4 to 9 [13]. The η isoform has exons 2–9 removed, and the ϕ isoform lacks exons 3 through 6. The ω isoform lacks exons 1–3 and encodes proteins of different lengths due to different translation starting sites in exons 4 and 5 [17]. The most recently identified isoforms are π, which has the C-terminal portion of exon 4 deleted, and κ, which lacks exon 3 and the N-terminal portion of exon 4 [14][18]. The RING domain of BARD1 is mapped to exons 2 and 3 [19]. Its ANK repeats span from the end of exon 4 to exon 7 while its BRCT domain encompasses exons 8–11. It is thought that the different isoforms may play a role in tumorigenesis through the disrupting of BARD1’s important protein-protein interactions. These variations have been well-studied in hereditary breast and ovarian cancers, however, the presence and functional consequences of these alterations in other cancer types are still being investigated.

Figure 2. BARD1 variants in non-breast and non-gynecological cancers (A) BARD1 isoforms. Full-length BARD1 (BARD1-FL) and different variants that occur due to alternative splicing. The table depicts in which cancer types the isoforms are oncogenic or tumor suppressors. * indicates multiple forms of alternative splicing. The γ isoform is either composed of only exons 1–3 or exons 1–3, 5–11. The ω1 isoform is depicted in this figure, which begins at a.a. 382. The ω2 isoform begins in exon 4 at a.a. 407 while the ω3 isoform begins in exon 7 at a.a. 538. NB: neuroblastoma. CRC: colorectal cancer. NSCLC: non-small cell lung cancer. AML: acute myeloid leukemia. (B) SNPs and genetic mutations in the BARD1 gene. BARD1 is composed of 11 exons (white) and introns (gray) and includes 5′ and 3′ UTR (black lines). Genetic variants of BARD1 in NB (red), CRC (blue), pancreatic cancer (green), nephroblastoma (pink), and Ewing sarcoma (purple) are depicted as lines localized to the region of the mutation. Only exons are drawn to scale.
3. BRCA1-Dependent Function of BARD1
3.1. BARD1-BRCA1 E3 Ubiquitin Ligase Activity
BARD1 and BRCA1 interact via the RING domains located at their respective N-termini. This interaction allows for the formation of a heterodimer with E3 ubiquitin ligase activity [20][21]. As an ubiquitin ligase, BARD1-BRCA1 promotes ubiquitination of targeted proteins and their proteasomal degradation; however, its atypical K6 ubiquitin linkages, though poorly understood, are thought to act in signal transduction [22][23][24]. BARD1-BRCA1 can even auto-ubiquitinate to increase its own activity levels and stability [22][25]. The heterodimer formation may be vital for the stabilization of BARD1 and BRCA1 as the loss of one protein drastically decreases the amount of the other protein [21].
Many of the heterodimer’s roles relate to facilitating mitosis. For example, BARD1-BRCA1 has been shown to localize to and ubiquitinate centrosome proteins, particularly γ-tubulin and the γ-tubulin ring complex, to inhibit microtubule nucleation at the centrosomes [26][27][28][29]. Loss of BARD1-BRCA1 at this step results in the rapid accumulation of fragmented or extra centrosomes [27][29]. Intriguingly, BRCA1, likely with the help of BARD1, can also promote DNA damage-induced centrosome amplification, possibly as a defense mechanism in response to prolonged DNA damage [30]. BARD1-BRCA1 ubiquitination activity is also required for TPX2, a spindle fiber organizer, to properly aggregate at the spindle poles [31]. Together, these studies confer both centrosome-dependent and -independent functions in spindle apparatus assembly during metaphase and anaphase. During telophase and cytokinesis, BARD1-BRCA1 ubiquitinates Aurora B kinase, a chromosomal segregation kinase, which leads to its turnover [32]. BARD1-BRCA1′s action against Aurora B is thought to confine it to the mitotic contractile ring. Interestingly, this likely occurs via Aurora B binding to TACC1 at the midbody with protection by BARD1β [3][32][33].
3.2. BARD1-BRCA1 in Homologous Recombination
BRCA1-BARD1 has long been implicated in homologous recombination (HR). Although the heterodimer has many biological functions, its role in HR is perhaps the most thoroughly investigated and likely contributes the most to its tumor-suppressing functions. Therein, the BARD1-BRCA1 complex has been primarily implicated in DNA end resection and presynaptic complex formation.
3.2.1. DNA End Resection
To commit to HR, two processes must occur. First, the 53BP1-RIF1-Shieldin complex, which aggregates to protect dsDNA ends after double-strand break (DSB) formation, needs to be removed [39][40]. The BARD1-BRCA1 heterodimer is thought to facilitate this process by ubiquitinating histone H2A. This enables SMARCAD1 recruitment, a chromatin remodeler, which facilitates the removal of the 53BP1-containing complex [36]. Second, the 5′ ends of DSBs are resected to generate 3′ overhangs of ssDNA. These overhangs then serve as docking sites for the assembly of pro-HR proteins and commit DNA repair to the HR pathway. CtIP binds the BRCT domain of BRCA1 as well as the MRE11-RAD50-NBS1 (MRN) complex [41][42]. Together with other nucleases, the MRN-CtIP complex processes the DSB ends and promotes HR [43]. BARD1-BRCA1 interacting with CtIP indicates a potential function in DNA end resection. In line with this, studies have demonstrated that BRCA1 improved resection speed [44]. However, in all, this process is not exclusively BRCA1-dependent [45][46].
3.2.2. Presynaptic Complex Formation
The presynaptic complex, or the presynaptic filament, refers to the RAD51-ssDNA nucleoprotein filament that is formed at the 3′ ends of resected DNA, which then searches and invades the homologous strand and forms a D-loop [47]. Immediately after DNA end resection, Replication Protein A (RPA) binds to and protects the ssDNA until RAD51 is loaded onto the ssDNA to replace them [48]. RAD51, a DNA recombinase, is a key catalyst for strand invasion, homology search, and pairing of DNA during HR. Both BARD1 and BRCA1 are capable of physically interacting with RAD51 and DNA; however, it is the BRCA2-DSS1 complex that facilitates the replacement of RPA with RAD51 [49]. PALB2 binds both BRCA1 and BRCA2 and functions as a bridge between them. BRCA1 and PALB2 interact via their respective coiled-coil domains [50]. Once bound to BRCA1, the PALB2′s WD40 domain enables the recruitment of BRCA2-DSS1 to DSBs [51]. BARD1-BRCA1-PALB2 effectively serves as a scaffold for BRCA2-mediated RAD51 loading. Additionally, the binding of BARD1-BRCA1 to RAD51 is thought to aggregate RAD51 for improved efficiency and success of RPA replacement by BRCA2-DSS1, but this idea has yet to be tested [52]. The formation of the BARD1-BRCA1-PALB2-BRCA2-DSS1 complex occurs in a stepwise fashion. Mutations therein alter DNA foci in a parallel stepwise manner with BRCA1 mutations impairing the complex from forming entirely [53]. Thus, BARD1-BRCA1 is central to the recruitment of BRCA2-DSS1 to DSBs in HR.
3.3. BARD1-BRCA1 in Mismatch Repair
Both BARD1 and BRCA1 have been shown to interact with MSH2 and MLH2, two important mismatch repair (MMR) proteins [54][55][56]. These molecular interactions suggest that BARD1-BRCA1 may contribute to MMR. However, evidence supporting this is conflicted, as genetic studies suggest a functional interaction between BRCA1 and MSH2 in HR [56]. Overall, BARD1-BRCA1 involvement in MMR is underexplored, but this connection could explain the incidence of colorectal cancer (CRC) in BRCA1- and BARD1-mutated CRC patients as MMR defects have long been known to predispose individuals to CRC [57].
4. BRCA1-Independent Functions of BARD1
4.1. Regulation of p53 and Apoptosis
BARD1 has been implicated in the regulation of apoptosis. BARD1 overexpression induces apoptosis, while the tumor-related mutation, Q564H, diminishes BARD1’s pro-apoptotic ability when exposed to genotoxic stress [58].
The function of BARD1-related apoptosis is associated with binding to p53, Ku70, and the phosphorylation of p53 at Serine-15 [15][58]. Binding of p53 occurs in the ANK-BRCT linker region and the BRCT domain [15][59][60]. In cell lines that are apoptosis-resistant and deficient in phosphorylated Serine-15 of p53 (NuTu-19 and HEK 293T cell lines), BARD1 overexpression can restore the phosphorylation capacity. This suggests that BARD1 promotes the formation of p53 and DNA-PK complexes, which then allows for p53 phosphorylation by ATM and results in apoptosis [15]. Additionally, mutations associated with breast, ovarian, and uterine malignancies lack sequences in the ANK-BRCT linker region that are vital for BARD1-dependent apoptosis [59]. BARD1 has also been shown to stabilize p53 in a slightly different setting. In cervical cancer progression, human papillomavirus (HPV) 16 E6 transforming protein aims to inactivate p53, however, BARD1 adds a layer of protection by increasing and stabilizing p53 while inactivating E6 [61].
BARD1 exerts its pro-apoptotic function in the cytoplasm after BARD1 is shuttled out of the nucleus [7][59]. Schüchner et al. discovered three NLS within BARD1 that are centrally located and do not overlap with other functional domains [8]. Just one NLS sequence is sufficient for translocating BARD1 into the nucleus, which is independent of BRCA1 [59]. BARD1 also contains a CRM1-dependent NES at the N-terminus following the RING domain, allowing for nuclear export through CRM1 [7]. Interestingly, BRCA1 suppresses BARD1 apoptotic activity by interfering with its export, shifting BARD1 towards its BRCA1-dependent cell survival activity [7][58]. Without the influences of BRCA1, BARD1 can localize to the cytoplasm and induce apoptosis [7][59].
Furthermore, Tembe et al. showed that BARD1-p53 localizes to the mitochondria where BARD1 disrupts the mitochondrial membrane potential and induces apoptosis [60][62]. BARD1 also has the ability to relocate to the mitochondria without p53, however, p53 likely acts as an intermediary in BARD1-related downregulation of Bcl-2 leading to Bax oligomerization [63]. BARD1’s ability to induce Bax oligomerization and apoptosis relies on an intact BRCT domain [60]. The deletion of its BRCT domain not only limits the ability of BARD1 to bind p53, but it also inhibits its export out of the nucleus and localization to the cytoplasm and mitochondria [60].
On the other hand, BARD1 promotes the import of BRCA1 to the nucleus, whereas p53 regulates BRCA1 export by interfering with BRCA1-BARD1 binding [63][64]. BRCA1, similar to BARD1, also induces apoptosis in the cytoplasm [7][60][64]. However, BARD1 inhibits the export of BRCA1 out of the nucleus and disrupts the ubiquitin E3 ligase activity, both of which disrupt BRCA1-dependent apoptosis [64]. Mutating the NLS of BARD1 results in BRCA1 localizing to the cytoplasm, thus supporting the hypothesis that BARD1 acts as a chaperone to transport BRCA1 into the nucleus [8].
4.2. Cell Cycle Regulation/Mitosis
The amount of BRCA1 protein increases during S-phase, while the level of BARD1 protein remains constant from G1 to G2 but increases during mitosis [65][66]. BARD1 does play a vital role in S-phase progression [66]. Cell cycle-dependent kinase complexes, CDK2-cyclin A1/E1 and CDK1-cyclin B, phosphorylate BARD1 resulting in potentiation of its function in mitosis [67].
Localization and overexpression of BARD1 in the nucleus, but not the cytoplasm, results in G1 cell cycle arrest [8]. Similar to the BARD1 location-dependent apoptosis described above, cell cycle regulation may be dependent on whether BARD1 is localized to the nucleus or the cytoplasm.
4.3. NF-κB
BARD1 can also bind NF-κB and regulate its transcriptional activity [68][69]. BARD1, in addition to Pirin and Tip60, forms a Bcl-3 interacting proteins (BIP) network that then forms a quaternary complex with Bcl-3 and p50 [68]. Specifically, from half of the ANK into the BRCT domains, BARD1 binds the ANK repeats of Bcl-3 [68]. The complex then interacts with the promoter of the NF-κB gene and activates its transcription. Regarding BRCA1, BARD1 has been shown to inhibit the ability of BRCA1 to activate transcription through NF-κB [70].
4.4. Inhibition of mRNA Processing in Response to DNA Damage
A polyadenylation factor, CstF-50, has been shown to bind BARD1 in the nucleus to inhibit polyadenylation of mRNA [71]. CstF-50 binds the ANK-BRCT linker region of BARD1, thus involving BARD1 in mRNA processing and stabilization of RNA polymerase II (RNAP II) in response to DNA damage [6][71][72]. Within the RNAP II holoenzyme, BARD1 can recognize sites of DNA damage, and, through inhibition of polyadenylation of mRNA, BARD1 acts to prevent the processing of immature transcripts that may otherwise be translated to deleterious proteins [71][73]. The inhibition of transcription is further ensured through BRCA1-BARD1 mediated ubiquitination of RNAP II [74]. In response to DNA damage, cells have decreased levels of polyadenylation with concurrent increases in the CstF-50-BARD1-BRCA1 complex [73]. BARD1 is phosphorylated by CDK-cyclin at Threonine-734, which is important for its interaction with CstF-50 [65][75]. The role of BARD1 in mRNA processing has implications in tumorigenesis. For example, the Q564H mutation of BARD1 found in ovarian, breast, and uterine tumors, reduces the BARD1-CstF-50 interaction, and prevents their inhibition of polyadenylation [73]. Interestingly, p53 associates with BARD1-CstF-50, and tumor-related mutations in p53 also result in decreased BARD1-CstF-50 association and inhibition of mRNA cleavage [76].
4.5. ADP Ribosylation, Poly (ADP-Ribose) Polymerase and BARD1
The transfer of ADP-ribose from NAD+ to a target protein is called ADP ribosylation. A group of enzymes, called poly (ADP-ribose) polymerases (PARPs), further catalyze the polymerization of ADP-ribose (PAR) [77]. These later reactions are termed PARsylation. Nuclear PARPs play a critical role in DDR and genome stability [78]. Inhibition of PARPs has been implemented in the treatment of BRCA1/2 mutated cancers [79][80][81][82][83]. BARD1 plays an essential role in the PAR signaling in response to DNA damage. Specifically, BARD1’s BRCT domain directly binds the ADP-ribose within PAR and recruits BRCA1 to the damage sites [79].
References
- Wu, L.C.; Wang, Z.W.; Tsan, J.T.; Spillman, M.A.; Phung, A.; Xu, X.L.; Yang, M.C.W.; Hwang, L.Y.; Bowcock, A.M.; Baer, R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 1996, 14, 430–440.
- Cimmino, F.; Formicola, D.; Capasso, M. Dualistic role of BARD1 in cancer. Genes 2017, 8, 375.
- Irminger-Finger, I.; Ratajska, M.; Pilyugin, M. New concepts on BARD1: Regulator of BRCA pathways and beyond. Int. J. Biochem. Cell Biol. 2016, 72, 1–17.
- Brzovic, P.S.; Rajagopal, P.; Hoyt, D.W.; King, M.C.; Klevit, R.E. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 2001, 8, 833–837.
- Birrane, G.; Varma, A.K.; Soni, A.; Ladias, J.A. Crystal structure of the BARD1 BRCT domains. Biochemistry 2007, 46, 7706–7712.
- Edwards, R.A.; Lee, M.S.; Tsutakawa, S.E.; Williams, R.S.; Tainer, J.A.; Glover, J.M. The BARD1 C-terminal domain structure and interactions with polyadenylation factor CstF-50. Biochemistry 2008, 47, 11446–11456.
- Rodriguez, J.A.; Schüchner, S.; Au, W.W.; Fabbro, M.; Henderson, B.R. Nuclear–cytoplasmic shuttling of BARD1 contributes to its proapoptotic activity and is regulated by dimerization with BRCA1. Oncogene 2004, 23, 1809–1820.
- Schüchner, S.; Tembe, V.; Rodriguez, J.A.; Henderson, B.R. Nuclear targeting and cell cycle regulatory function of human BARD1. J. Biol. Chem. 2005, 280, 8855–8861.
- Fox, D.; Le Trong, I.; Rajagopal, P.; Brzovic, P.S.; Stenkamp, R.E.; Klevit, R.E. Crystal structure of the BARD1 ankyrin repeat domain and its functional consequences. J. Biol. Chem. 2008, 283, 21179–21186.
- Thai, T.H.; Du, F.; Tsan, J.T.; Jin, Y.; Phung, A.; Spillman, M.A.; Massa, H.F.; Muller, C.Y.; Ashfaq, R.; Michael Mathis, J.; et al. Mutations in the BRCA1-associated RING domain (BARD1) gene in primary breast, ovarian and uterine cancers. Hum. Mol. Genet. 1998, 7, 195–202.
- Karppinen, S.M.; Heikkinen, K.; Rapakko, K.; Winqvist, R. Mutation screening of the BARD1 gene: Evidence for involvement of the Cys557Ser allele in hereditary susceptibility to breast cancer. J. Med. Genet. 2004, 41, e114.
- Ghimenti, C.; Sensi, E.; Presciuttini, S.; Brunetti, I.M.; Conte, P.; Bevilacqua, G.; Caligo, M.A. Germline mutations of the BRCA1-associated ring domain (BARD1) gene in breast and breast/ovarian families negative for BRCA1 and BRCA2 alterations. Genes Chromosom. Cancer 2002, 33, 235–242.
- Li, L.; Ryser, S.; Dizin, E.; Pils, D.; Krainer, M.; Jefford, C.E.; Bertoni, F.; Zeillinger, R.; Irminger-Finger, I. Oncogenic BARD1 isoforms expressed in gynecological cancers. Cancer Res. 2007, 67, 11876–11885.
- Zhang, Y.Q.; Pilyugin, M.; Kuester, D.; Leoni, V.P.; Li, L.; Casula, G.; Zorcolo, L.; Schneider-Stock, R.; Atzori, L.; Irminger-Finger, I. Expression of oncogenic BARD1 isoforms affects colon cancer progression and correlates with clinical outcome. Br. J. Cancer 2012, 107, 675–683.
- Feki, A.; Jefford, C.E.; Berardi, P.; Wu, J.Y.; Cartier, L.; Krause, K.H.; Irminger-Finger, I. BARD1 induces apoptosis by catalysing phosphorylation of p53 by DNA-damage response kinase. Oncogene 2005, 24, 3726–3736.
- Tsuzuki, M.; Wu, W.; Nishikawa, H.; Hayami, R.; Oyake, D.; Yabuki, Y.; Fukuda, M.; Ohta, T. A truncated splice variant of human BARD1 that lacks the RING finger and ankyrin repeats. Cancer Lett. 2006, 233, 108–116.
- Lepore, I.; Dell’Aversana, C.; Pilyugin, M.; Conte, M.; Nebbioso, A.; De Bellis, F.; Tambaro, F.P.; Izzo, T.; Garcia-Manero, G.; Ferrara, F.; et al. HDAC inhibitors repress BARD1 isoform expression in acute myeloid leukemia cells via activation of miR-19a and/or b. PLoS ONE 2013, 8, e83018.
- Zhang, Y.Q.; Bianco, A.; Malkinson, A.M.; Leoni, V.P.; Frau, G.; De Rosa, N.; André, P.A.; Versace, R.; Boulvain, M.; Laurent, G.J.; et al. BARD1: An independent predictor of survival in non-small cell lung cancer. Int. J. Cancer 2012, 131, 83–94.
- Ratajska, M.; Matusiak, M.; Kuzniacka, A.; Wasag, B.; Brozek, I.; Biernat, W.; Koczkowska, M.; Debniak, J.; Sniadecki, M.; Kozlowski, P.; et al. Cancer predisposing BARD1 mutations affect exon skipping and are associated with overexpression of specific BARD1 isoforms. Oncol. Rep. 2015, 34, 2609–2617.
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, H.; Ohta, T. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem. 2001, 276, 14537–14540.
- Xia, Y.; Pao, G.M.; Chen, H.W.; Verma, I.M.; Hunter, T. Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J. Biol. Chem. 2003, 278, 5255–5263.
- Wu-Baer, F.; Lagrazon, K.; Yuan, W.; Baer, R. The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J. Biol. Chem. 2003, 278, 34743–34746.
- Nishikawa, H.; Ooka, S.; Sato, K.; Arima, K.; Okamoto, J.; Klevit, R.E.; Fukuda, M.; Ohta, T. Mass spectrometric and mutational analyses reveal Lys-6-linked polyubiquitin chains catalyzed by BRCA1-BARD1 ubiquitin ligase. J. Biol. Chem. 2004, 279, 3916–3924.
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422.
- Chen, A.; Kleiman, F.E.; Manley, J.L.; Ouchi, T.; Pan, Z.Q. Autoubiquitination of the BRCA1*BARD1 RING ubiquitin ligase. J. Biol. Chem. 2002, 277, 22085–22092.
- Hsu, L.C.; Doan, T.P.; White, R.L. Identification of a gamma-tubulin-binding domain in BRCA1. Cancer Res. 2001, 61, 7713–7718.
- Starita, L.M.; Machida, Y.; Sankaran, S.; Elias, J.E.; Griffin, K.; Schlegel, B.P.; Gygi, S.P.; Parvin, J.D. BRCA1-dependent ubiquitination of gamma-tubulin regulates centrosome number. Mol. Cell Biol. 2004, 24, 8457–8466.
- Sankaran, S.; Starita, L.M.; Groen, A.C.; Ko, M.J.; Parvin, J.D. Centrosomal microtubule nucleation activity is inhibited by BRCA1-dependent ubiquitination. Mol. Cell Biol. 2005, 25, 8656–8668.
- Sankaran, S.; Crone, D.E.; Palazzo, R.E.; Parvin, J.D. BRCA1 regulates gamma-tubulin binding to centrosomes. Cancer Biol. Ther. 2007, 6, 1853–1857.
- Zou, J.; Zhang, D.; Qin, G.; Chen, X.; Wang, H.; Zhang, D. BRCA1 and FancJ cooperatively promote interstrand crosslinker induced centrosome amplification through the activation of polo-like kinase 1. Cell Cycle 2014, 13, 3685–3697.
- Joukov, V.; Groen, A.C.; Prokhorova, T.; Gerson, R.; White, E.; Rodriguez, A.; Walter, J.C.; Livingston, D.M. The BRCA1/BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell 2006, 127, 539–552.
- Ryser, S.; Dizin, E.; Jefford, C.E.; Delaval, B.; Gagos, S.; Christodoulidou, A.; Krause, K.H.; Birnbaum, D.; Irminger-Finger, I. Distinct roles of BARD1 isoforms in mitosis: Full-length BARD1 mediates Aurora B degradation, cancer-associated BARD1beta scaffolds Aurora B and BRCA2. Cancer Res. 2009, 69, 1125–1134.
- Delaval, B.; Ferrand, A.; Conte, N.; Larroque, C.; Hernandez-Verdun, D.; Prigent, C.; Birnbaum, D. Aurora B-TACC1 protein complex in cytokinesis. Oncogene 2004, 23, 4516–4522.
- Thakar, A.; Parvin, J.; Zlatanova, J. BRCA1/BARD1 E3 ubiquitin ligase can modify histones H2A and H2B in the nucleosome particle. J. Biomol. Struct. Dyn. 2010, 27, 399–406.
- Zhu, Q.; Pao, G.M.; Huynh, A.M.; Suh, H.; Tonnu, N.; Nederlof, P.M.; Gage, F.H.; Verma, I.M. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 2011, 477, 179–184.
- Densham, R.M.; Garvin, A.J.; Stone, H.R.; Strachan, J.; Baldock, R.A.; Daza-Martin, M.; Fletcher, A.; Blair-Reid, S.; Beesley, J.; Johal, B.; et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol. 2016, 23, 647–655.
- Stewart, M.D.; Zelin, E.; Dhall, A.; Walsh, T.; Upadhyay, E.; Corn, J.E.; Chatterjee, C.; King, M.C.; Klevit, R.E. BARD1 is necessary for ubiquitylation of nucleosomal histone H2A and for transcriptional regulation of estrogen metabolism genes. Proc. Natl. Acad. Sci. USA 2018, 115, 1316–1321.
- Calvo, V.; Beato, M. BRCA1 counteracts progesterone action by ubiquitination leading to progesterone receptor degradation and epigenetic silencing of target promoters. Cancer Res. 2011, 71, 3422–3431.
- Noordermeer, S.M. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121.
- Dev, H.; Chiang, T.W.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965.
- Yu, X.; Wu, L.C.; Bowcock, A.M.; Aronheim, A.; Baer, R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J. Biol. Chem. 1998, 273, 25388–25392.
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514.
- Anand, R.; Ranjha, L.; Cannavo, E.; Cejka, P. Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell 2016, 64, 940–950.
- Cruz-Garcia, A.; Lopez-Saavedra, A.; Huertas, P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 2014, 9, 451–459.
- Reczek, C.R.; Szabolcs, M.; Stark, J.M.; Ludwig, T.; Baer, R. The interaction between CtIP and BRCA1 is not essential for resection-mediated DNA repair or tumor suppression. J. Cell Biol. 2013, 201, 693–707.
- Polato, F.; Callen, E.; Wong, N.; Faryabi, R.; Bunting, S.; Chen, H.T.; Kozak, M.; Kruhlak, M.J.; Reczek, C.R.; Lee, W.H.; et al. CtIP-mediated resection is essential for viability and can operate independently of BRCA1. J. Exp. Med. 2014, 211, 1027–1036.
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257.
- Bhat, K.P.; Cortez, D. RPA and RAD51: Fork reversal, fork protection, and genome stability. Nat. Struct. Mol. Biol. 2018, 25, 446–453.
- Zhao, W.; Steinfeld, J.B.; Liang, F.; Chen, X.; Maranon, D.G.; Ma, C.J.; Kwon, Y.; Rao, T.; Wang, W.; Sheng, C.; et al. BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 2017, 550, 360–365.
- Sy, S.M.; Huen, M.S.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160.
- Oliver, A.W.; Swift, S.; Lord, C.J.; Ashworth, A.; Pearl, L.H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009, 9, 990–996.
- Zhao, W.; Wiese, C.; Kwon, Y.; Hromas, R.; Sung, P. The BRCA Tumor Suppressor Network in Chromosome Damage Repair by Homologous Recombination. Annu. Rev. Biochem. 2019, 88, 221–245.
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600.
- Wang, Q.; Zhang, H.; Guerrette, S.; Chen, J.; Mazurek, A.; Wilson, T.; Slupianek, A.; Skorski, T.; Fishel, R.; Greene, M.I. Adenosine nucleotide modulates the physical interaction between hMSH2 and BRCA1. Oncogene 2001, 20, 4640–4649.
- Romeo, F.; Falbo, L.; Di Sanzo, M.; Misaggi, R.; Faniello, M.C.; Viglietto, G.; Cuda, G.; Costanzo, F.; Quaresima, B. BRCA1 is required for hMLH1 stabilization following doxorubicin-induced DNA damage. Int. J. Biochem. Cell Biol. 2011, 43, 1754–1763.
- Maresca, L.; Spugnesi, L.; Lodovichi, S.; Cozzani, C.; Naccarato, A.G.; Tancredi, M.; Collavoli, A.; Falaschi, E.; Rossetti, E.; Aretini, P.; et al. MSH2 role in BRCA1-driven tumorigenesis: A preliminary study in yeast and in human tumors from BRCA1-VUS carriers. Eur. J. Med. Genet. 2015, 58, 531–539.
- Phelan, C.M.; Iqbal, J.; Lynch, H.T.; Lubinski, J.; Gronwald, J.; Moller, P.; Ghadirian, P.; Foulkes, W.D.; Armel, S.; Eisen, A.; et al. Incidence of colorectal cancer in BRCA1 and BRCA2 mutation carriers: Results from a follow-up study. Br. J. Cancer 2014, 110, 530–534.
- Irminger-Finger, I.; Leung, W.C.; Li, J.; Dubois-Dauphin, M.; Harb, J.; Feki, A.; Jefford, C.E.; Soriano, J.V.; Jaconi, M.; Montesano, R.; et al. Identification of BARD1 as mediator between proapoptotic stress and p53-dependent apoptosis. Mol. Cell 2001, 8, 1255–1266.
- Jefford, C.E.; Feki, A.; Harb, J.; Krause, K.H.; Irminger-Finger, I. Nuclear–cytoplasmic translocation of BARD1 is linked to its apoptotic activity. Oncogene 2004, 23, 3509–3520.
- Tembe, V.; Martino-Echarri, E.; Marzec, K.A.; Mok, M.T.; Brodie, K.M.; Mills, K.; Lei, Y.; DeFazio, A.; Rizos, H.; Kettle, E.; et al. The BARD1 BRCT domain contributes to p53 binding, cytoplasmic and mitochondrial localization, and apoptotic function. Cell. Signal. 2015, 27, 1763–1771.
- Yim, E.-K.; Lee, K.-H.; Myeong, J.; Tong, S.-Y.; Um, S.J.; Park, J.-S. Novel interaction between HPV E6 and BARD1 (BRCA1-associated ring domain 1) and its biologic roles. DNA Cell Biol. 2007, 26, 753–761.
- Tembe, V.; Henderson, B.R. BARD1 translocation to mitochondria correlates with Bax oligomerization, loss of mitochondrial membrane potential, and apoptosis. J. Biol. Chem. 2007, 282, 20513–20522.
- Jiang, J.; Yang, E.S.; Jiang, G.; Nowsheen, S.; Wang, H.; Wang, T.; Wang, Y.; Billheimer, D.; Chakravarthy, A.B.; Brown, M.; et al. p53-dependent BRCA1 nuclear export controls cellular susceptibility to DNA damage. Cancer Res. 2011, 71, 5546–5557.
- Fabbro, M.; Savage, K.; Hobson, K.; Deans, A.J.; Powell, S.N.; McArthur, G.A.; Khanna, K.K. BRCA1-BARD1 complexes are required for p53Ser-15 phosphorylation and a G1/S arrest following ionizing radiation-induced DNA damage. J. Biol. Chem. 2004, 279, 31251–31258.
- Choudhury, A.D.; Xu, H.; Baer, R. Ubiquitination and proteasomal degradation of the BRCA1 tumor suppressor is regulated during cell cycle progression. J. Biol. Chem. 2004, 279, 33909–33918.
- Irminger-Finger, I.; Soriano, J.V.; Vaudan, G.; Montesano, R.; Sappino, A.P. In vitro repression of Brca1-associated RING domain gene, Bard1, induces phenotypic changes in mammary epithelial cells. J. Cell Biol. 1998, 143, 1329–1339.
- Hayami, R.; Sato, K.; Wu, W.; Nishikawa, T.; Hiroi, J.; Ohtani-Kaneko, R.; Fukuda, M.; Ohta, T. Down-regulation of BRCA1-BARD1 ubiquitin ligase by CDK2. Cancer Res. 2005, 65, 6–10.
- Dechend, R.; Hirano, F.; Lehmann, K.; Heissmeyer, V.; Ansieau, S.; Wulczyn, F.G.; Scheidereit, C.; Leutz, A. The Bcl-3 oncoprotein acts as a bridging factor between NF-κB/Rel and nuclear co-regulators. Oncogene 1999, 18, 3316–3323.
- Irminger-Finger, I.; Leung, W.-C. BRCA1-dependent and independent functions of BARD1. Int. J. Biochem. Cell Biol. 2002, 34, 582–587.
- Benezra, M.; Chevallier, N.; Morrison, D.J.; MacLachlan, T.K.; El-Deiry, W.S.; Licht, J.D. BRCA1 augments transcription by the NF-κB transcription factor by binding to the Rel domain of the p65/RelA subunit. J. Biol. Chem. 2003, 278, 26333–26341.
- Kleiman, F.E.; Manley, J.L. Functional interaction of BRCA1-associated BARD1 with polyadenylation factor CstF-50. Science 1999, 285, 1576–1579.
- Cevher, M.A.; Kleiman, F.E. Connections between 3’-end processing and DNA damage response. Wiley Interdiscip. Rev. RNA 2010, 1, 193–199.
- Kleiman, F.E.; Manley, J.L. The BARD1-CstF-50 interaction links mRNA 3’ end formation to DNA damage and tumor suppression. Cell 2001, 104, 743–753.
- Kleiman, F.E.; Wu-Baer, F.; Fonseca, D.; Kaneko, S.; Baer, R.; Manley, J.L. BRCA1/BARD1 inhibition of mRNA 3’ processing involves targeted degradation of RNA polymerase II. Genes Dev. 2005, 19, 1227–1237.
- Kim, H.-S.; Li, H.; Cevher, M.; Parmelee, A.; Fonseca, D.; Kleiman, F.E.; Lee, S.B. DNA Damage–Induced BARD1 Phosphorylation Is Critical for the Inhibition of Messenger RNA Processing by BRCA1/BARD1 Complex. Cancer Res. 2006, 66, 4561–4565.
- Nazeer, F.I.; Devany, E.; Mohammed, S.; Fonseca, D.; Akukwe, B.; Taveras, C.; Kleiman, F.E. p53 inhibits mRNA 3’ processing through its interaction with the CstF/BARD1 complex. Oncogene 2011, 30, 3073–3083.
- Hottiger, M.O. Poly(ADP-ribose) polymerase inhibitor therapeutic effect: Are we just scratching the surface? Expert Opin. Ther. Targets 2015, 19, 1149–1152.
- Azarm, K.; Smith, S. Nuclear PARPs and genome integrity. Genes Dev. 2020, 34, 285–301.
- Li, M.; Yu, X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 2013, 23, 693–704.
- Bürkle, A. Physiology and pathophysiology of poly (ADP-ribosyl) ation. Bioessays 2001, 23, 795–806.
- Fisher, A.E.; Hochegger, H.; Takeda, S.; Caldecott, K.W. Poly (ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly (ADP-ribose) glycohydrolase. Mol. Cell. Biol. 2007, 27, 5597–5605.
- Okano, S.; Lan, L.; Caldecott, K.W.; Mori, T.; Yasui, A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol. Cell. Biol. 2003, 23, 3974–3981.
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; De Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447.

