Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jerome Moreaux | + 6159 word(s) | 6159 | 2021-08-16 04:51:11 | | | |
| 2 | Vivi Li | Meta information modification | 6159 | 2021-09-26 03:23:01 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Moreaux, J. Transcription/Replication Conflicts in Myelomagenesis. Encyclopedia. Available online: https://encyclopedia.pub/entry/14535 (accessed on 27 June 2026).
Moreaux J. Transcription/Replication Conflicts in Myelomagenesis. Encyclopedia. Available at: https://encyclopedia.pub/entry/14535. Accessed June 27, 2026.
Moreaux, Jerome. "Transcription/Replication Conflicts in Myelomagenesis" Encyclopedia, https://encyclopedia.pub/entry/14535 (accessed June 27, 2026).
Moreaux, J. (2021, September 24). Transcription/Replication Conflicts in Myelomagenesis. In Encyclopedia. https://encyclopedia.pub/entry/14535
Moreaux, Jerome. "Transcription/Replication Conflicts in Myelomagenesis." Encyclopedia. Web. 24 September, 2021.
Copy Citation
Multiple myeloma is a hematologic cancer characterized by the accumulation of malignant plasma cells in the bone marrow. It remains a mostly incurable disease due to the inability to overcome refractory disease and drug-resistant relapse. Oncogenic transformation of PC in multiple myeloma is thought to occur within the secondary lymphoid organs. However, the precise molecular events leading to myelomagenesis remain obscure. Here, we identified genes involved in the prevention and the resolution of conflicts between the replication and transcription significantly overexpressed during the plasma cell differentiation process and in multiple myeloma cells. We discussed the potential role of these factors in myelomagenesis.
multiple myeloma
transcription replication conflicts
R-loops
G-quadruplexes
genomic instability
tumorigenesis
plasma cells
1. Introduction
Human plasma cells (PCs) represent the final stage of B lymphocyte differentiation and play an essential role in the humoral immune response by secreting antibodies. They are mainly located in the bone marrow, where they normally represent only 0.25% of all bone marrow mononuclear cells [1]. In lymph nodes, native B cells are induced to become either memory B cells (MBCs) or plasmablastic cells. In the latter case, plasmablastic cells migrate rapidly to the bone marrow niches or other tissues where they find the adequate microenvironment for long-term survival [2][3]. Mature PCs are characterized by very high immunoglobulin (Ig) secretion.
In adult life, B lymphocytes are continuously produced, and during their differentiation in either MBC or towards PCs, they undergo several genetic rearrangements, associated with DNA breaks to finally assure the immense variability in Igs: VDJ recombination, Ig class switch recombination (CSR), and somatic hyper-mutation (SHM). CSR and SHM take place in the germinal center of the secondary lymphoid organs. These events need to be tightly regulated to ensure an efficient immune response without auto-immune reactions, and to prevent tumorigenesis. This requires adequate processing of the physiological R-loops occurring in guanine-rich switch (S) regions of the immunoglobulin heavy chain (IgH) locus. R-loops are three-stranded nucleic acid structures, formed by the annealing of an RNA moiety with double-stranded DNA constituting an RNA:DNA hybrid [4]. These structures are physiologically enriched near promoters and transcription termination sites, and are involved in CSR, transcription initiation and termination, and telomere elongation [5]. In contrast, unscheduled R-loop formation interferes with replication fork progression and increases the collision rate between the replication and transcription machineries [6], known as transcription/replication conflicts (TRCs) [7]. Therefore, R-loops not only facilitate programmed recombination such as CSR but also represent an important source of spontaneous genomic instability, and their formation must be tightly regulated to prevent tumorigenesis [8][9][10].
Our group developed a multi-step cell culture system to model B-cell to PC differentiation where various combinations of cytokines and activation molecules are needed to reproduce the sequential PC differentiation steps occurring in the different organs/tissue in vivo [11][12][13]. MBCs differentiate into pre-plasmablasts (prePBs), plasmablasts (PBs), early PCs, and finally, long-lived PCs that produce high Ig amounts [11][12][13]. PrePBs have been identified in human lymph nodes, tonsils, and bone marrow [12][14]. This transitional stage is characterized by the absence of both B-cell and PC markers. In particular, at this stage, DNA replication and transcription rates dramatically increase since prePBs start to produce large amounts of Igs and still have a high proliferation rate [12][15], a unique situation possibly prone to give rise to increased TRCs. Therefore, a tight regulation between transcription and replication at this step is critical to avoid unscheduled DNA breaks that might lead to oncogenic transformation. Using our mRNA expression data that are available at ArrayExpress (http://www.ebi.ac.uk/arrayexpress/ accessed on 23 July 2021, E-MTAB-1771, E-MEXP-2360 and E-MEXP-3034), we found that several factors and protein complexes involved in TRC prevention or resolution are overexpressed in prePBs (Figure 1), highlighting the importance of mechanisms required for proper TRC processing during human PC differentiation. Interestingly, several of these factors are overexpressed in malignant PCs from patients with multiple myeloma (MM) compared with PCs from healthy donors. Among these factors, XRN2, DDX1, DDX23 HNRNPU, HNRNPD, SRPK2 and SRSF1 have been identified in the R-loop interactome, reinforcing their role in R-loop biology [16]. Multiple myeloma is an incurable hematological cancer characterized by malignant PC accumulation in the bone marrow. These cells are characterized by high genomic instability, due to oncogene-induced replication stress, and cell cycle deregulation [9][17][18]. Genomic abnormalities in MM involve somatic mutations and translocations between an oncogene (MMSET, CCND3, CCND1, MAF) and the open chromatin of a topologically-associated domain (TAD) of an IgH locus. As mentioned above, since malignant PCs produce high Ig amounts while they keep dividing, an ever increasing efficiency of TRC resolution might confer a significant selective advantage during myelomagenesis and disease progression. Therefore, promoting TRC persistence could constitute an interesting new therapeutic strategy to treat MM.
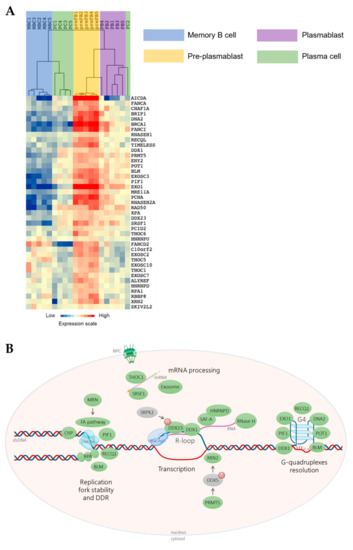
Figure 1. Transcription/replication conflicts (TRCs) resolution genes expression during memory B cells (MBCs) differentiation. (A) The genes significantly overexpressed in pre-plasmablasts (prePBs) compared to MBCs, PBs and plasma cells (PCs) were determined with a SAM (significance analysis of microarrays) multiclass analysis (false discovery rate (FDR) = 0), identifying 41 unique genes. When a gene was assayed by several probe sets, the probe set with the highest variance was used. An unsupervised hierarchical clustering was run on this list of 41 unique genes. The normalized expression value for each gene is indicated by a color, with red representing high expression and blue representing low expression. (B) TRCs resolution machinery. Green: factor overexpressed in prePBs. Grey: non-overexpressed factor. M: methylation. P: phosphorylation. S: SUMOylation. Simple arrow: activation or recruitment. Double arrow: interaction. dsDNA: double-strand DNA. G4: G-quadruplex. NPC: nuclear pore complex. The scheme was made using BioRender.
2. Management of Transcription/Replication Conflicts (TRCs) in Normal Cells Is Critical to Prevent Genomic Instability
During normal PC differentiation, the prePB stage is associated with a high rate of cell proliferation induced after B-cell activation, with around 50% of cells in S phase. Besides, Ig secretion starts at this stage resulting in strong Ig gene transcription [12][15]. PrePBs are hence exposed to both transcriptional and replication stresses that could enhance the occurrence of DNA lesions due to collisions between replication and transcription machineries. The role of TRCs in cancer has been significantly studied [19][20], however the specific involvement of this process in PC tumorigenesis have never been described. We hypothesized that, particularly at the prePB stage, TRCs have to be carefully managed. First, we sought to identify genes involved in TRC resolution that are significantly overexpressed in prePBs during B-cell to PC differentiation. By reviewing the existing literature, we identified 83 genes involved in TRC resolution (Table S1). Then, we quantified their expression during PC differentiation using our in vitro model and Affymetrix microarray data (data are available at ArrayExpress; http://www.ebi.ac.uk/arrayexpress/ accessed on 23 July 2021, E-MTAB-1771, E-MEXP-2360 and E-MEXP-3034). GenomicScape webtool [21] and multi-class SAM (significance analysis of microarrays) analysis revealed that 41 of these genes were significantly overexpressed in prePBs compared with MBCs, PBs and PCs, with a false discovery rate <5% (Figure 1 and Table S2). Among them, we focused on 38 genes that are described hereafter.
2.1. R-Loop Resolution Genes
2.1.1. RNase H1/2, Replication Protein A (RPA)
Ribonucleases H (RNase H) comprise a group of enzymes that degrade the RNA strand of RNA:DNA hybrids. There are two main classes of RNases H in eukaryotic cells, distinguished by their primary sequences and substrate specificities [22][23]. RNase H1 is a monomeric enzyme that resolves long RNA:DNA hybrids and removes the ribonucleotides in DNA that are at least four nucleotides long [22]. In mammalian cells, RNase H1 is essential for R-loop processing during mitochondrial DNA replication and its deletion causes embryonic lethality in mice [24][25][26]. RNase H1 depletion also resulted in increased nuclear RNA:DNA hybrids, DNA damage and slowing of DNA replication forks [27], implying that RNase H1 plays an important role in resolving TRCs in the nucleus.
RNase H2 is a heterotrimeric complex that processes hybrids and removes single ribonucleotide in DNA. Ribonucleotides are incorporated during genome replication at a remarkably high rate [28][29]. These mis-incorporated nucleotides in nascent DNA need to be efficiently removed by the ribonucleotide excision repair (RER) pathway, a process similar to Okazaki fragment processing after incision by RNase H2 [30]. Biallelic mutations in RNase H2 are linked to a neuroinflammatory disease, Aicardi-Goutières syndrome, presumably through the accumulation of cytosolic DNA fragments and the activation of the DNA sensing cGAS/STING pathway [31][32]. RNase H2 plays, therefore, a critical role in the maintenance of genome integrity. During normal PC differentiation, the prePB stage is associated with high cell proliferation following B-cell activation with about half the cell population in the S phase. Increased expression of RNase H2 in prePBs could be critical to reduce R-loop levels and maintain genome integrity (Figure 1A).
RPA is a major single-stranded DNA (ssDNA) binding protein conserved in all eukaryotes [33][34]. It has been shown that RPA colocalizes with R-loops. In vitro, RPA stimulates RNase H1 activity on R-loops. Moreover, RPA-RNase H1 interaction is critical for RNase H1 binding to R-loops [35]. Importantly, RPA interacts with various TRC resolution factors that will be described below and, therefore, is a key regulator of TRC-induced genomic instability. As RPA is involved also in replication fork progression and restart, its function is not restricted to prevent unscheduled R-loop formation [36]. Importantly, it has been shown that the proteasome inhibitor Bortezomib, used in the treatment of MM, prevents DNA resection and thus RPA recruitment onto ssDNA [37][38]. Moreover, higher RPA expression is associated with an increased bone marrow infiltration of MM cells which is associated with a poor outcome [39].
2.1.2. The DEAD-Box Protein Family of Helicases
The DEAD-box RNA helicase 1 (DDX1) is involved in RNA metabolism [40][41] and DNA double strand break (DSB) repair [42][43]. At DSB sites, DDX1 forms foci to resolve DNA:RNA hybrids [42][43], and could also be involved in G-quadruplex (G4) structures remodeling [44]. G4s are four-stranded secondary DNA structures, constituted of at least two stacked guanine tetrads stabilized by Hoogsteen hydrogen bonds and cations [45][46]. These highly stable non-canonical structures are present at telomeres, at the promoter of many genes, and at origins replication [47][48]. A link between G4 structures and R-loops has been proposed since they are both promoted by Guanine-richness (G-richness), and G4s can form in the displaced DNA strand of a R-loop and stabilize it [49][50]. Moreover, a recent study demonstrated that in human cells, genomic instability caused by G4 stabilizers is mediated by R-loop formation [51], reinforcing the interplay between these structures. Interestingly, DDX1 could promote CSR by a mechanism involving G4 structures conversion into R-loops [44]. Long non-coding RNAs (lncRNAs) are transcribed from the intronic S region of the IgH locus (called germline transcripts), and are involved in the formation of co-transcriptional R-loops during CSR that allows the activity of the key CSR enzyme, namely Activation-Induced cytidine Deaminase (AID) [52][53]. Ribeiro de Almeida et al. described a post-transcriptional mechanism by which G4s form in these lncRNAs after their splicing and are subsequently recognize by both AID and DDX1. The latter would promote the conversion of G4 RNAs into RNA:DNA hybrids at the S region, thereby allowing AID targeting and activity. The authors showed that CSR is impaired in Ddx1 knock-out mice, and DDX1 depletion reduces R-loop levels at IgH S regions [44].
Another DEAD-box RNA helicase, DDX23, is part of the U5 spliceosomal ribonucleoprotein particle (U5 snRNP) involved in messenger RNA (mRNA) splicing, depending on its phosphorylation by SRPK2 [54]. A recent study proposed that the accumulation of R-loops induces RNA polymerase II (RNA Pol II) pausing which will recruit DDX23 to resolve these R-loops [55]. This process requires DDX23 phosphorylation by SRPK2. Upon depletion of both factors, R-loop levels and genomic instability are enhanced.
2.2. mRNA Maturation
2.2.1. Serine/Arginine Splicing Factor 1 (SRSF1)
SRSF1 (or ASF/SF2) is an mRNA splicing factor that belongs to the serine/arginine-rich family [56][57][58]. SRSF1-depleted cells are hyper-mutagenic, with DNA rearrangements and increased DSB formation, and their genetic instability is R-loop-dependent. In vitro, SRSF1 associates with RNA Pol II to prevent R-loop formation, and it has recently been shown that SRSF1 depletion in human cells increases R-loop levels [59][60]. Moreover, SRSF1 is required for the SUMOylation of Topoisomerase I (Top1) [61], an enzyme that relieves torsional stress during transcription and replication [62][63]. The SUMOylation is a post-translational modification performed by a member of the Small Ubiquitin-Like Modifier (SUMO) family such as SUMO1. This modification allows the interaction of Top1 with RNA Pol II at actively transcribed genes, and the recruitment of mRNA processing factors preventing R-loop formation. Importantly, the authors showed that Top1 SUMOylation reduces its catalytic activity meaning that the prevention of R-loops might not involve supercoiled DNA relaxation. Instead, they hypothesized that this inhibition could prevent the formation of trapped Top1-DNA complexes that create genomic instability [61][64].
2.2.2. Heterogeneous Nuclear Ribonucleoprotein U (HNRNPU) and D0 (HNRNPD)
HNRNPU (or SAF-A) is an RNA processing factor involved in transcription elongation, RNA stability, and splicing [65][66][67]. HNRNPU is transiently recruited to DNA damaged sites and then rapidly released from chromatin, suggesting a role in DNA repair [68]. Its release depends on transcription and on the activity of the three PI3-kinase-related protein kinase (PIKK) controlling the DNA damage response (DDR), namely ATM, ATR and DNA PK [69]. Upon inhibition of these kinases, HNRNPU remains on chromatin, and R-loop levels are increased, suggesting that its release is important for R-loop resolution at DNA damaged sites.
A recent study showed that HNRNPD interacts with HNRNPU [70]. HNRNPD regulates DDR genes and acts to preserve genomic integrity [71]. It was shown that HNRNPD is necessary for proper DNA end resection during homologous recombination and for the removal of R-loops [70]. HNRNPD depletion reduces HNRNPU recruitment to damaged sites and induces R-loop accumulation. Importantly, a study showed that ILF2 is critical in the pathophysiology of MM cells with 1q21 amplification. They showed that ILF2 interacts with HNRNP U/D among other RNA-binding proteins and modulates the splicing of DNA repair genes [72].
Interestingly, FANCD2 recruits HNRNPU and the DDX47 helicase under replication stress, and HNRNPU or DDX47 depletion leads to R-loop accumulation [73].
2.2.3. The THO/TREX Complex and TREX2
The THO-TREX complex participates in messenger ribonucleoprotein (mRNP) biogenesis and mRNA export [74][75]. This complex has also been involved in preventing transcription-associated genomic instability [76]. Indeed, yeast THO mutants display growth defects, and require a functional S-phase checkpoint for survival upon R-loop accumulation [77]. In these mutants, transcriptional downregulation is observed genome-wide, but particularly at long, highly expressed, and G-rich genes. Interestingly, the recruitment of Rrm3 (a PIF-family helicase that promotes replication progression across DNA obstacles) to transcribed genes is enhanced in THO-depleted mutants, and this recruitment is RNase H-sensitive [78]. Gomez-Gonzalez et al. thus propose that THO acts genome-wide to prevent transcription-dependent replication defects related to R-loop formation.
In human cells, THO depletion impairs transcription elongation, increases DNA damage and spontaneous recombination in a RNase H-sensitive manner [79]. Of note, AID overexpression in THO-depleted murine B cells increases genomic instability and CSR. Indeed, AID catalyzes the deamination of a cytosine in the displaced DNA strand of a R-loop, that will be recognized and processed by DNA repair mechanisms leading to DNA breaks. Replication is altered in THO-depleted cells and the authors discussed the possibility that defects in replication termination or inactivation of DNA damage checkpoints would lead to longer replication tracks. They also hypothesized that the impairment of transcription elongation in those cells would reduce transcription activity and promote the progression of the replication fork. Moreover, THO associates with the DEAD-box RNA helicase UAP56 (DDX39B) that was shown to prevent R-loop-mediated genomic instability and unwind R-loops in vitro [75][80].
Interestingly, a recent paper showed that the Sin3A histone deacetylase (HDAC) interacts with THOC1, a member of the THO complex [81]. The yeast homolog of Sin3A suppresses R-loop-dependent genomic instability, and in human cells genomic instability induced by Sin3A depletion can be rescued by RNase H-overexpression. In Sin3A-depleted cells as well as in THOC1-depleted cells, histone acetylation is enhanced and upon incubation with a HDAC inhibitor, R-loop levels are increased. It is most likely that chromatin opening due to increased acetylation facilitates R-loop formation. This can also explain why in THO-depleted cells, replication forks are faster, but fork pausing or stalling occurs more frequently [81]. Finally, a recent study showed that THOC1 depletion in hepatocellular carcinoma cells induces R-loop accumulation and increases sensitivity to cisplatin [82].
The THSC-TREX2 complex is involved in mRNA export, interacts with the nuclear pore complex [83][84][85], and has a role in alleviating transcription-associated DNA damage. TREX2 yeast mutants display high level of transcription-associated hyperrecombination and downregulation of long, highly transcribed, and G-rich genes, as observed in THO mutants. The two main components of the TREX2 complex, Thp1 (PCID2) and Sac3 (GANP), act as a complex and bind to highly transcribed genomic regions to which THO also binds. Moreover, in yeast TREX2 mutants, TRCs are increased and replication forks stall at Sac3 and Thp1 binding sites. However, the direct involvement of R-loops in this process remains elusive [86]. Another study on TREX2 role in TRC resolution in human cells [87] found that TREX2 depletion increases genomic instability and DNA damage. Of note, the homologous recombination factor BRCA2 interacts with two TREX2 components, PCID2 and DSS1, and upon BRCA2 depletion the accumulation of R-loops is observed [87]. However, the precise role of TREX2 in TRC and R-loop resolution is still unclear.
2.3. RNA Processing and Degradation
2.3.1. The RNA Exosome
The RNA exosome is a ribonucleolytic complex involved in RNA processing and degradation [88][89], and is composed of nine non-catalytic subunits (EXOSC1-EXOSC9) and two catalytic subunits (EXOSC10 and DIS3). DIS3 mutations are involved in MM progression [90]. Indeed, DIS3 mutations are associated with a poor prognosis and are associated with significant transcriptional changes [91][92]. Moreover, germline variants in DIS3 were identified in familial MM [93]. A recent study demonstrated that EXOSC10 degrades DNA damage-induced long non-coding RNAs that are synthesized at DSB sites and might be involved in R-loop formation [94]. Moreover, the RNA exosome cooperates with the helicase Senataxin (SETX) for R-loop removal, and EXOSC9 colocalizes with SETX in an R-loop-dependent manner [95]. SETX or RNA exosome depletion in B cells increases genomic instability and impairs CSR [96][97]. SETX could resolve R-loops and recruit the RNA exosome for RNA degradation. Optimal activity of the RNA exosome complex has been shown to be mandatory for clearing non-coding RNAs from R-loops formed at S regions, thus facilitating the occurrence of cytidine deamination by AID on both strands of R-loops for optimal CSR [98]. The specific catabolism of non-coding RNAs by the DIS3 subunit was additionally shown to impact TAD structures genome-wide [99].
2.3.2. 5′-3′ Exoribonuclease 2 (XRN2)
XRN2 is involved in transcription termination [100][101]. XRN2 could link transcription and DNA repair because upon DNA damage, it forms foci that colocalize with several DDR factors. Of note, XRN2 colocalizes with R-loops upon UV exposure in a transcription-dependent manner, and XRN2 depletion increases R-loop and DSB levels [102]. It has been shown recently that XRN2 resolves RNA:DNA hybrids to allow the initiation of DNA repair by non-homologous end-joining (NHEJ) [103]. XRN2 could work together with several factors for R-loop resolution. For example, the R-loop unwinding activity of SETX allows XRN2 access to RNA in order to degrade it [104]. Moreover, XRN2 interacts with the DDX5 helicase [105][106], and this interaction requires DDX5 arginine methylation by PRMT5 [107]. One hypothesis is that DDX5 unwinds RNA-DNA hybrids and that subsequently XRN2 degrades the RNA moiety. Upon depletion of PRMT5, DDX5, or XRN2, R-loops accumulate at highly transcribed genes [108]. Interestingly, high PRMT5 expression is associated with an adverse outcome in MM [109]. Moreover, PRMT5 depletion or inhibition in MM cells inhibits cell growth and induce apoptosis in association with NFκB pathway downregulation [109].
2.4. Fork Protection and Stability
2.4.1. The Fanconi Anemia Pathway and the MRN Complex
Fanconi anemia (FA) is a rare genetic disorder characterized by congenital abnormalities as well as an increased susceptibility to cancers and to hematopoietic failure [110][111][112]. The best characterized function of the FA pathway is the removal of inter-strand crosslinks and subsequent DNA repair by HR [113][114]. The FA pathway has also been implicated in R-loop resolution. Specifically, two studies demonstrated that FANCD2 is required for efficient R-loop resolution the prevention of genomic instability [115][116]. Interestingly, high levels of FANCD2 expression are associated with shorter survival in MM [117]. Upon FANCD2 depletion, DSBs accumulate at R-loop sites in a transcription- and R-loop-dependent manner. When DNA replication is inhibited using aphidicolin, a DNA polymerase inhibitor, FANCD2 accumulates at large transcribed genes and colocalizes with R-loops in a transcription-dependent manner [115][116]. Another study demonstrated that FANCD2 participates in R-loop elimination at common fragile sites to allow their replication. FANCD2 depletion increases the R-loop-dependent genomic instability at these sites [118]. Monoubiquitylation of the FANCI-FANCD2 complex is required for R-loop resolution [119], and FANCI-FANCD2 binding to the displaced DNA strand or to the RNA tail of the R-loop stimulates its monoubiquitylation. FANCD2 recruitment to chromatin is promoted by transcription R-loop and DNA damage [119]. In FA-deficient cells, R-loops accumulate at some loci to which the FA core complex protein FANCA binds, and FANCD2 foci are sensitive to RNase H1 treatment [115]. Moreover, the DEAD/DEAH helicase FANCM might participate in RNA:DNA hybrid unwinding, at least in vitro, and its depletion induces R-loop accumulation [116]. Therefore, the FA pathway plays a significant role in R-loop resolution and in genomic instability prevention, through several effectors. Interestingly, FANCI is part of a gene prognostic signature in MM patients [120]. FANCA depletion has been identified in a CRISPR-Cas9 screen to sensitize MM cells to melphalan [121].
A recent study also demonstrated that the MRN (MRE11, RAD50, NBS1/NBN) complex has a role in promoting R-loop resolution by the FA pathway [122], and that this role is independent from its catalytic activity. The authors showed that the MRN complex is required for FANCD2 and FANCM recruitment to R-loops. In MRN-depleted cells, R-loop accumulation and R-loop-dependent DNA damage are increased. MRE11 and RAD50 expression levels correlate with high bone marrow infiltration in MM [39].
The FA pathway could also contribute to G4 resolution. Indeed, the DNA helicase FANCJ (BRIP1) can bind to and unwind G4 structures in vitro [123][124][125]. FANCJ depletion increases sensitivity to the G4 stabilizing molecule telomestatin suggesting a role in preventing G4-induced genomic instability [123]. Moreover, cells derived from patients with FA and FANCJ deficiency accumulate large genomic deletions in G4-prone regions, reinforcing the protective role of FANCJ against DNA damage induced by G4 structures [124]. In Xenopus laevis egg extracts, treatment with G4 stabilizers promotes RNA Pol II stalling and increases the FANCJ requirement to ensure the efficient replication of G4-containing regions [126]. Accordingly, FANCJ bypasses G4 structures in vitro and unwinds downstream DNA to allow its faithful replication [125]. Importantly, cancer-associated FANCJ mutations [127] reduce its ability to unwind G4 structures and increase sensitivity to G4 stabilizing agents [128].
Finally, a recent study reported that FANCJ interacts with the helicase REV1 via a PCNA-interaction peptide (PIP)-like motif to form a G4-resolving complex [129]. REV1 binds preferentially to G4 DNA substrates in vitro, prevents G4 folding, and disrupts G4 DNA structures [130]. Moreover, its depletion could increase the mutational rate at G4 DNA sites [131].
2.4.2. Breast Cancer Susceptibility Gene 1 and 2 (BRCA1 and BRCA2)
BRCA1 and BRCA2 are part of the FA pathway [132]. Specifically, BRCA1 is a tumor suppressor with an extensively documented role in DNA repair and homologous recombination (HR) [133]. In MM, the FA/BRCA pathway contributes to melphalan resistance and targeting this pathway can potentiate the response to melphalan treatment [134][135]. Importantly, the NFκB pathway frequently deregulated in MM is known to promote HR through stimulation of BRCA1 and CtIP [136][137]. BRCA1 is recruited to R-loops and forms a complex with the SETX helicase to suppress co-transcriptional and R-loop-induced DNA damage. Moreover, BRCA1 is associated with transcription termination sites of highly transcribed genes that are enriched in genomic alterations in BRCA1-deficent breast tumors [138]. Of note, R-loop accumulation is observed upon BRCA1 knock-down in human cells [87][139] and in BRCA1 mutation-carrying precancerous breast tissue [140]. Moreover, R-loops preferentially accumulate at loci associated with RNA Pol II pausing in these cells [140].
A recent study showed that BRCA2 associates with the DEAD-box RNA helicase DDX5, which resolves RNA:DNA hybrids at DSBs to facilitate DNA repair [105][106][141][142]. Their interaction is enhanced upon DNA damage and reduced by RNase H1 overexpression. BRCA2 depletion leads to R-loop accumulation at DSBs and promotes DDX5 recruitment to DNA damage sites, presumably to facilitate homologous recombination [141]. Moreover, BRCA2 can stimulate DDX5 unwinding activity in vitro [141]. Importantly, DDX5 interaction with the BRCA2 mutant T207A, found in breast cancer cells from patients, is reduced compared to wild-type BRCA2, and R-loops levels are increased in cells that overexpress this variant [141].
2.4.3. Proliferating Cell Nuclear Antigen (PCNA) SUMOylation
PCNA, one of the main components of the replication fork, enhances DNA polymerase processivity and is involved in DDR and genome stability maintenance [143]. In MM, PCNA expression increases with disease progression [144][145]. PCNA targeting induces apoptosis and increases the efficacy of several treatments including melphalan, doxorubicine, thalidomide and azacitidine [146]. PCNA can be conjugated to SUMO1 to prevent homologous recombination at DSBs [147][148], and conjugation with SUMO2 has recently been involved in TRC resolution [149]. Indeed, PCNA conjugation to SUMO2 on transcribed genes during S phase positively regulates replication fork progression, in a RECQ5-dependent manner. This conjugation destabilizes RNA Pol II binding, thus reducing transcription and facilitating replication [149]. Interestingly, SUMO2-conjugated PCNA interacts with the histone chaperone CAF1, and enhances CAF1-dependent histone deposition, thereby forming repressive chromatin [149]. Upon SUMO2-PCNA conjugation abrogation, TRCs and DNA damage are increased. The helicase RECQ5 allows the interaction between PCNA and RNA Pol II, and between PCNA and SUMO2, and suppresses TRC-induced DSBs through chromatin remodeling [149]. Interestingly, SUMO2 could be involved in induction of bortezomib resistance upon silencing of Sentrin/SUMO-specific proteases-2 (SENP2) [150].
2.4.4. CtBP-Interacting Protein (CtIP)
CtIP (RBBP8) is a 5′ flap endonuclease a regulator of MRN activity, critical for DNA end resection at DSBs [151][152][153]. CtIP expression levels are associated with relapse and with a poor prognosis in MM [154]. A recent study showed a role for CtIP in R-loop processing [155]. The authors described a mechanism by which CtIP would recognize and process 5′ flaps that are present inside the R-loop structure, to promote the activity of helicases that would remove the RNA strand and therefore resolve the R-loop structure. Accordingly, CtIP-depleted cells show more R-loops but fewer DNA breaks. CtIP depletion in both human and yeast cells reduces their survival upon exposition to the Top1 inhibitor camptothecin (CPT), and increases R-loop formation in a transcription-dependent manner. Moreover, in CtIP-depleted cells exposed to CPT, transcription patterns are altered mainly for R-loop-prone genes, and SETX chromatin binding is increased. R-loop resolution by CtIP requires its nuclease activity and is observed in both untreated and CPT-treated cells. Intriguingly, concomitant loss of CtIP and of the XPG nucleotide excision repair endonuclease abrogates R-loop formation, whereas depletion of each protein on its own promotes R-loop formation. In yeast, depletion of the CtIP homolog Sae2 increases RNA Pol II stalling upon CPT exposure specifically during S-phase [155].
2.4.5. Exonuclease 1 (EXO1)
EXO1 is a 5′-3′ exonuclease involved in mismatch repair and DSB resection during homologous recombination [156]. EXO1 depletion in human cells leads to spontaneous telomere defects, to the stalling of replication forks preferentially at G4 structures and enhances the cell sensitivity to G4 stabilizers. EXO1 could have a protective role on the replication fork that stall in front of a G4 structure, by resecting the nascent DNA and promoting repair by homologous recombination. Accordingly, EXO1-depleted cells display less resection around G4 structures and the collapsed forks are mainly repaired by the error-prone NHEJ repair pathway [157].
2.4.6. Transcription Coupled Nucleotide Excision Repair (TC-NER) Exonucleases
Structure-specific endonucleases, such as XPF and XPG, recognize and process specific secondary DNA structures to facilitate replication or DNA repair [158]. As mentioned before, it is thought that R-loop structures contain at least two 5′ flaps that could be recognized by such enzymes [155]. XPF and XPG process R-loop structures formed in the switch region of the IgH locus in vitro [159]. In human cells, XPG and XPF can process R-loops induced by depletion of the RNA processing factor Aquarius (AQR), in a nuclease activity-dependent manner [160]. Moreover, XPG cleaves R-loops that accumulate upon depletion of various R-loop resolution factors (AQR, SETX, or SRSF1) or upon CPT exposure. This mechanism requires other components of the transcription-coupled nucleotide excision repair (TC-NER) such as XPA, XPB, XPD and CSB. XPF can bind to R-loops and is enriched at R-loop sites of gene bodies upon cell exposure with CPT, and can induce ssDNA breaks within R-loops [161]. Moreover, XPG activity is necessary for RAD52-dependent R-loop resolution [162]. It has been hypothesized that the ability of TC-NER components to process R-loops could be a way to distinguish between physiological and unscheduled R-loops, by acting only on gene bodies R-loops that are detrimental for the replication fork progression [163]. In MM, shorter overall survival is associated with single nucleotide polymorphisms (SNPs) in XPG and XPA genes [164]. Additionally, adult T-cell leukemia cells accumulate R-loops and often lack TC-NER factors, such as XPF and XPG [165].
2.5. G-Quadruplexes Resolution
2.5.1. The RecQ family of Helicases
Bloom’s syndrome helicase (BLM) is a DNA helicase of the RecQ family [166] involved in replication fork restart, notably via G4 unwinding [167][168][169]. BLM-depleted cells are characterized by increased genomic instability, DNA damage, sister chromatid exchanges (SCE) and micronuclei formation [170][171]. Upon BLM depletion, genes enriched in putative G4 sequences are downregulated, suggesting that the G4 unwinding activity of BLM plays an additional role in transcription regulation [172][173]. Moreover, in BLM-depleted cells, SCEs occur mostly at G4 motif-containing sites in actively transcribed genes [171]. Interestingly, the phenotype of BLM-depleted cells can be reversed by RNase H overexpression, suggesting that R-loops are involved in the phenotypes of BLM deficiency. BLM colocalizes with R-loops, and its depletion leads to R-loop accumulation [170]. Sgs1, the yeast homolog of BLM, can reduce R-loop formation and DNA damage levels at fragile sites and R-loop-prone genomic loci. Sgs1-depleted cells are characterized by increased R-loop levels, transcription-associated recombination, and DNA damage. This phenotype is exacerbated when Sgs1 loss is combined with the loss of another TRC resolution gene (TREX, RNase H, or the Senataxin homolog Sen1) [170]. Altogether, these data show that BLM is a crucial helicase that can act on both G4s and R-loops to reduce DNA damage formation.
Moreover, the HERC2 E3 ligase can enhance BLM unwinding activity, presumably through promoting the interaction of BLM with RPA. HERC2 promotes this interaction mainly during S phase and its E3 ligase activity is required for BLM helicase activity. This could involve HERC2-mediated phosphorylation and ubiquitination of RPA. HERC2-depleted cells display increased G4 formation and SCEs occurrence. They are sensitized to G4 stabilizing molecules, such as pyridostatin and telomestatin [174][175]. Finally, HERC2 E3 ligase activity has an epistatic relationship with RPA regarding G4 resolution [176]. Therefore, HERC2 is an important regulator of G4 resolution.
Additionally, a study demonstrated the direct interaction between Top1 and BLM. It was shown in vitro that Top1 stimulates BLM unwinding activity on RNA:DNA hybrids, and reciprocally, BLM stimulates Top1 activity on DNA:RNA hybrids, in a helicase activity-independent manner [177]. More recently, RECQ DNA helicase BLM was shown to be overexpressed in prePBs during B to PC differentiation [178]. BLM could restrain the deleterious consequences of R-loop mediated replication stress in highly proliferative prePB cells upon activation of transcription. In the absence of BLM, these cells would, therefore, accumulate stalled forks leading to chromosome breaks due to their inability to efficiently remove R-loops and G4 structures [178].
RECQ1 is an ATP-dependent DNA helicase that has a role in genome maintenance and DDR. Its depletion increases chromosomal breaks and sensitizes cells to replication blocking agents [179][180]. Like BLM, RECQ1 helicase activity is also stimulated by RPA [181]. Moreover, in MM cells, RECQ1 depletion induces and accumulation of cells in G1 and G2/M phases and increased apoptosis, whereas RECQ1 overexpression protects these cells from bortezomib or melphalan-induced cell death [182]. Melphalan is an alkylating agent classically used in the treatment of MM. Moreover, RECQ1 mRNA levels are upregulated upon DNA damage in a p53-dependent manner [183]. Interestingly, genes downregulated upon RECQ1 depletion are enriched in G4 motifs, and RECQ1 can bind to G4 structures at their promoter [184][185]. Of note, RECQ1 interacts with several members of the TREX1 complex, suggesting that it could cooperate with this complex in TRC resolution [182]. However, RECQ1 cannot unwind G4 structures on its own in vitro [168]. Though, RECQ1 might contribute to G4 motif-related DNA repair, since RECQ1 is rapidly recruited to oxidized chromatin. Additionally, it has been shown that the guanine residues present in G4 motif-containing promoters are prone to oxidization, forming an 8-oxoguanine lesion [186]. In line with this hypothesis, a recent study showed that PARP1, a sensor of DNA damage, is recruited to oxidized G4s and that this interaction can promote PARP1 activation. This could lead to G4s sensing and signaling to recruit DNA repair enzymes for G4 removal [187]. RECQ1 interacts with PARP1and its depletion re-sensitizes MM cells to PARP inhibitors [182], reinforcing the hypothesis of RECQ1 being involved in G4-related DNA repair. However, RECQ1 role in G4 resolution needs to be clarified.
2.5.2. PIF1
The DNA helicases of the PIF1 family are conserved among eukaryotes. This family comprises two members in budding yeast, Pif1 and Rrm3, whereas in most eukaryotes only PIF1 is present [188]. PIF1 regulates telomerase activity and Okazaki fragment maturation [188]. PIF1 can unwind G4s in vitro [189][190][191]. In budding yeast, it is necessary for the efficient replication of G4-containing sequences [192][193][194]. In the absence of Pif1, replication fork progression is slower. Moreover, Pif1 interaction with PCNA is crucial to allow G4-forming sequences to be replicated [192]. PIF1 and RPA have complementary roles in DNA replication across G4 sequences [195] in vitro, with RPA allowing replication through G4 DNA, and PIF1 unwinding these structures. Their interaction is DNA-dependent [196]. RPA unwinds G4s in vitro [197][198], prevents genomic instability at G-rich motifs, and is crucial for G4 removal [196][199]. Human and yeast RPA prevents G4 formation in telomeres to allow telomere elongation [200].
Interestingly, Pif1 preferentially unwind RNA:DNA hybrids rather than DNA duplexes [201]. Moreover, Pif1 has a role in promoting replication through transfer RNA genes (tDNAs). These structures are enriched in R-loops, and the association of Pif1 with tDNAs is enhanced by RNase H depletion [202]. Therefore, Pif1 could have an additional role in R-loop resolution.
2.5.3. DNA2 and POT1 Roles at Telomeres
DNA2 is a helicase/nuclease involved in the maintenance of genomic stability [203]. Interestingly, it could have a redundant role with the Pif1 helicase at telomeres [204]. Indeed, yeast and human DNA2 bind to telomeric G4 DNA and can cleave these structures in an RPA-dependent manner [205][206]. DNA2 deficiency induces strong telomeric DNA damage [206]. Importantly, DNA2 is found throughout replicating DNA during S phase, but not on telomeres [207], and DNA2-depleted cells are characterized by high genomic instability (chromosome bridges, aneuploidy, replication defects) [208][209]. Moreover, DNA2 reduces replication stress in cancer cells [209]. Mouse DNA2 homozygous knock-out is embryonic lethal, demonstrating an essential role of this protein [206]. Interestingly, DNA2 interacts with PCNA and FANCD2 that are both involved in TRCs resolution (see above). It stimulates BLM DNA unwinding activity [210], but it is not known whether it also stimulates G4 unwinding.
POT1 is a component of the shelterin complex in charge of telomere protection and binds to the G-rich ssDNA 3′ overhang in telomeres [211]. In vitro, POT1 disrupts G4 formation at telomeres to allow proper elongation by telomerase [212]. The two G4 stabilizing molecules telomestatin and pyridostatin inhibits POT1 binding to telomeres in vitro and in human cells [175][213][214]. POT1 unfolds G4 DNA to produce ssDNA-POT1 complexes [215][216]. However, in ciliates, the POT1 homolog TEBPα (Telomere end-binding protein alpha) participate in the formation of telomeric G4s [217]. Interestingly, POT1 expression is associated with clinical characteristics related to adverse outcome in MM [218].
Despite the lack of studies involving all these factors specifically in malignant transformation of PCs, we hypothesized that the significant overexpression of TRCs resolution genes in prePBs might play an important role during memory B-cell to PC differentiation by preventing genomic instability and thus tumorigenesis. Under normal conditions, only a limited number of memory B cells are able to form new germinal center (GC) after reactivation. During early tumorigenesis, founder mutations acquired by memory B cells as SHM off-targets or resulting from DNA replication errors following B-cell activation can jeopardize this mechanism and yield a set of aberrant memory B cells that progressively outcompete wild-type memory B and naïve B cells along their clonal expansion [219]. Furthermore, participation in successive GC reactions is predicted to result in cumulative acquisition of further off-target mutations in these cells [219]. A recent study investigating the chronological activity of mutational signatures in MM, using a large cohort of 89 whole genome and 973 exome data, estimated that the transformation of a GC B cell occurred during the first second or third decades of life [220]. Furthermore, their data indicated that AID activity is not restricted to the first GC reaction but persists in at least a subset of patients, potentially affecting disease evolution. This supports that pre-malignant MM cells have re-entered the GC for clonal expansion decades before MM diagnosis [220]. Additional levels of genomic instability, related to AID-independent processes are also likely and could be related to replicative stress. Since pre-plasmablastic stage is characterized by high proliferation (50% of cells in S phase) and the beginning of Ig secretion [12], conflicts between transcription (likely mainly at the Ig loci) and replication during PC differentiation impact on replication stress and mutagenesis and might be involved in myelomagenesis. However, the presence of R-loops and G-quadruplexes at myeloma-associated mutation hotspots in prePBs remains to be demonstrated together with the mechanisms protecting them from genomic instability.
References
- Klein, B.; Tarte, K.; Jourdan, M.; Mathouk, K.; Moreaux, J.; Jourdan, E.; Legouffe, E.; De Vos, J.; Rossi, J.F. Survival and Proliferation Factors of Normal and Malignant Plasma Cells. Int. J. Hematol. 2003, 78, 106–113.
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The Generation of Antibody-Secreting Plasma Cells. Nat. Rev. Immunol. 2015, 15, 160–171.
- Cyster, J.G. Homing of Antibody Secreting Cells. Immunol. Rev. 2003, 194, 48–60.
- Hamperl, S.; Cimprich, K.A. The Contribution of Co-Transcriptional RNA:DNA Hybrid Structures to DNA Damage and Genome Instability. DNA Repair 2014, 19, 84–94.
- Sollier, J.; Cimprich, K.A. R-Loops Breaking Bad. Trends Cell Biol. 2015, 25, 514–522.
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-Loop-Mediated Genomic Instability Is Caused by Impairment of Replication Fork Progression. Genes Dev. 2011, 25, 2041–2056.
- Hamperl, S.; Cimprich, K.A. Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell 2016, 167, 1455–1467.
- Niehrs, C.; Luke, B. Regulatory R-Loops as Facilitators of Gene Expression and Genome Stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178.
- Alagpulinsa, D.A.; Szalat, R.E.; Poznansky, M.C.; Reis, R.J.S. Genomic Instability in Multiple Myeloma. Trends Cancer 2020, 6, 858–873.
- Hoang, P.H.; Cornish, A.J.; Chubb, D.; Jackson, G.; Kaiser, M.; Houlston, R.S. Impact of Mitochondrial DNA Mutations in Multiple Myeloma. Blood Cancer J. 2020, 10.
- Jourdan, M.; Caraux, A.; De Vos, J.; Fiol, G.; Larroque, M.; Cognot, C.; Bret, C.; Duperray, C.; Hose, D.; Klein, B. An in vitro Model of Differentiation of Memory B Cells into Plasmablasts and Plasma Cells Including Detailed Phenotypic and Molecular Characterization. Blood 2009, 114, 5173–5181.
- Jourdan, M.; Caraux, A.; Caron, G.; Robert, N.; Fiol, G.; Rème, T.; Bolloré, K.; Vendrell, J.-P.; Gallou, S.L.; Mourcin, F.; et al. Characterization of a Transitional Preplasmablast Population in the Process of Human B Cell to Plasma Cell Differentiation. J. Immunol. 2011, 187, 3931–3941.
- Jourdan, M.; Cren, M.; Robert, N.; Bolloré, K.; Fest, T.; Duperray, C.; Guilloton, F.; Hose, D.; Tarte, K.; Klein, B. IL-6 Supports the Generation of Human Long-Lived Plasma Cells in Combination with Either APRIL or Stromal Cell-Soluble Factors. Leukemia 2014, 28, 1647–1656.
- Leung-Hagesteijn, C.; Erdmann, N.; Cheung, G.; Keats, J.J.; Stewart, A.K.; Reece, D.E.; Chung, K.C.; Tiedemann, R.E. Xbp1s-Negative Tumor B Cells and Pre-Plasmablasts Mediate Therapeutic Proteasome Inhibitor Resistance in Multiple Myeloma. Cancer Cell 2013, 24, 289–304.
- Herviou, L.; Jourdan, M.; Martinez, A.-M.; Cavalli, G.; Moreaux, J. EZH2 Is Overexpressed in Transitional Preplasmablasts and Is Involved in Human Plasma Cell Differentiation. Leukemia 2019, 33, 2047–2060.
- Cristini, A.; Groh, M.; Kristiansen, M.S.; Gromak, N. RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell Rep. 2018, 23, 1891–1905.
- Bergsagel, P.L.; Kuehl, W.M.; Zhan, F.; Sawyer, J.; Barlogie, B.; Shaughnessy, J. Cyclin D Dysregulation: An Early and Unifying Pathogenic Event in Multiple Myeloma. Blood 2005, 106, 296–303.
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic Complexity of Multiple Myeloma and Its Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113.
- García-Muse, T.; Aguilera, A. Transcription–Replication Conflicts: How They Occur and How They Are Resolved. Nat. Rev. Mol. Cell Biol. 2016, 17, 553–563.
- Gómez-González, B.; Aguilera, A. Transcription-Mediated Replication Hindrance: A Major Driver of Genome Instability. Genes Dev. 2019, 33, 1008–1026.
- Kassambara, A.; Rème, T.; Jourdan, M.; Fest, T.; Hose, D.; Tarte, K.; Klein, B. GenomicScape: An Easy-to-Use Web Tool for Gene Expression Data Analysis. Application to Investigate the Molecular Events in the Differentiation of B Cells into Plasma Cells. PLoS Comput. Biol. 2015, 11.
- Cerritelli, S.M.; Crouch, R.J. Ribonuclease H: The Enzymes in Eukaryotes. FEBS J. 2009, 276, 1494–1505.
- Hyjek, M.; Figiel, M.; Nowotny, M. RNases H: Structure and Mechanism. DNA Repair 2019, 84, 102672.
- Ruhanen, H.; Ushakov, K.; Yasukawa, T. Involvement of DNA Ligase III and Ribonuclease H1 in Mitochondrial DNA Replication in Cultured Human Cells. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 2000.
- Lima, W.F.; Murray, H.M.; Damle, S.S.; Hart, C.E.; Hung, G.; De Hoyos, C.L.; Liang, X.-H.; Crooke, S.T. Viable RNaseH1 Knockout Mice Show RNaseH1 Is Essential for R Loop Processing, Mitochondrial and Liver Function. Nucleic Acids Res. 2016, 44, 5299–5312.
- Cerritelli, S.M.; Frolova, E.G.; Feng, C.; Grinberg, A.; Love, P.E.; Crouch, R.J. Failure to Produce Mitochondrial DNA Results in Embryonic Lethality in Rnaseh1 Null Mice. Mol. Cell 2003, 11, 807–815.
- Parajuli, S.; Teasley, D.C.; Murali, B.; Jackson, J.; Vindigni, A.; Stewart, S.A. Human Ribonuclease H1 Resolves R-Loops and Thereby Enables Progression of the DNA Replication Fork. J. Biol. Chem. 2017, 292, 15216–15224.
- Rydberg, B.; Game, J. Excision of Misincorporated Ribonucleotides in DNA by RNase H (Type 2) and FEN-1 in Cell-Free Extracts. Proc. Natl. Acad. Sci. USA 2002, 99, 16654–16659.
- Nick McElhinny, S.A.; Watts, B.E.; Kumar, D.; Watt, D.L.; Lundström, E.-B.; Burgers, P.M.J.; Johansson, E.; Chabes, A.; Kunkel, T.A. Abundant Ribonucleotide Incorporation into DNA by Yeast Replicative Polymerases. Proc. Natl. Acad. Sci. USA 2010, 107, 4949–4954.
- Zhou, Z.-X.; Williams, J.S.; Lujan, S.A.; Kunkel, T.A. Ribonucleotide Incorporation into DNA during DNA Replication and Its Consequences. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 109–124.
- Crow, Y.J.; Leitch, A.; Hayward, B.E.; Garner, A.; Parmar, R.; Griffith, E.; Ali, M.; Semple, C.; Aicardi, J.; Babul-Hirji, R.; et al. Mutations in Genes Encoding Ribonuclease H2 Subunits Cause Aicardi-Goutières Syndrome and Mimic Congenital Viral Brain Infection. Nat. Genet. 2006, 38, 910–916.
- Mackenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. CGAS Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature 2017, 548, 461–465.
- Wold, M.S. Replication Protein A: A Heterotrimeric, Single-Stranded DNA-Binding Protein Required for Eukaryotic DNA Metabolism. Annu. Rev. Biochem. 1997, 66, 61–92.
- Iftode, C.; Daniely, Y.; Borowiec, J.A. Replication Protein A (RPA): The Eukaryotic SSB. Crit. Rev. Biochem. Mol. Biol. 1999, 34, 141–180.
- Nguyen, H.D.; Yadav, T.; Giri, S.; Saez, B.; Graubert, T.A.; Zou, L. Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1. Mol. Cell 2017, 65, 832–847.e4.
- Mazina, O.M.; Somarowthu, S.; Kadyrova, L.Y.; Baranovskiy, A.G.; Tahirov, T.H.; Kadyrov, F.A.; Mazin, A.V. Replication Protein A Binds RNA and Promotes R-Loop Formation. J. Biol. Chem. 2020, 295, 14203–14213.
- Murakawa, Y.; Sonoda, E.; Barber, L.J.; Zeng, W.; Yokomori, K.; Kimura, H.; Niimi, A.; Lehmann, A.; Zhao, G.Y.; Hochegger, H.; et al. Inhibitors of the Proteasome Suppress Homologous DNA Recombination in Mammalian Cells. Cancer Res. 2007, 67, 8536–8543.
- Jacquemont, C.; Taniguchi, T. Proteasome Function Is Required for DNA Damage Response and Fanconi Anemia Pathway Activation. Cancer Res. 2007, 67, 7395–7405.
- Panero, J.; Stella, F.; Schutz, N.; Fantl, D.B.; Slavutsky, I. Differential Expression of Non-Shelterin Genes Associated with High Telomerase Levels and Telomere Shortening in Plasma Cell Disorders. PLoS ONE 2015, 10, e0137972.
- Godbout, R.; Li, L.; Liu, R.-Z.; Roy, K. Role of DEAD Box 1 in Retinoblastoma and Neuroblastoma. Future Oncol. 2007, 3, 575–587.
- Germain, D.R.; Graham, K.; Glubrecht, D.D.; Hugh, J.C.; Mackey, J.R.; Godbout, R. DEAD Box 1: A Novel and Independent Prognostic Marker for Early Recurrence in Breast Cancer. Breast Cancer Res. Treat. 2011, 127, 53–63.
- Li, L.; Monckton, E.A.; Godbout, R. A Role for DEAD Box 1 at DNA Double-Strand Breaks. Mol. Cell. Biol. 2008, 28, 6413–6425.
- Li, L.; Germain, D.R.; Poon, H.-Y.; Hildebrandt, M.R.; Monckton, E.A.; McDonald, D.; Hendzel, M.J.; Godbout, R. DEAD Box 1 Facilitates Removal of RNA and Homologous Recombination at DNA Double-Strand Breaks. Mol. Cell. Biol. 2016, 36, 2794–2810.
- Ribeiro de Almeida, C.; Dhir, S.; Dhir, A.; Moghaddam, A.E.; Sattentau, Q.; Meinhart, A.; Proudfoot, N.J. RNA Helicase DDX1 Converts RNA G-Quadruplex Structures into R-Loops to Promote IgH Class Switch Recombination. Mol. Cell 2018, 70, 650–662.e8.
- Hänsel-Hertsch, R.; Di Antonio, M.; Balasubramanian, S. DNA G-Quadruplexes in the Human Genome: Detection, Functions and Therapeutic Potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284.
- Rhodes, D.; Lipps, H.J. G-Quadruplexes and Their Regulatory Roles in Biology. Nucleic Acids Res. 2015, 43, 8627–8637.
- Prioleau, M.-N. G-Quadruplexes and DNA Replication Origins. Adv. Exp. Med. Biol. 2017, 1042, 273–286.
- Prorok, P.; Artufel, M.; Aze, A.; Coulombe, P.; Peiffer, I.; Lacroix, L.; Guédin, A.; Mergny, J.-L.; Damaschke, J.; Schepers, A.; et al. Involvement of G-Quadruplex Regions in Mammalian Replication Origin Activity. Nat. Commun. 2019, 10, 3274.
- Kuznetsov, V.A.; Bondarenko, V.; Wongsurawat, T.; Yenamandra, S.P.; Jenjaroenpun, P. Toward Predictive R-Loop Computational Biology: Genome-Scale Prediction of R-Loops Reveals Their Association with Complex Promoter Structures, G-Quadruplexes and Transcriptionally Active Enhancers. Nucleic Acids Res. 2018, 46, 7566–7585.
- Duquette, M.L. Intracellular Transcription of G-Rich DNAs Induces Formation of G-Loops, Novel Structures Containing G4 DNA. Genes Dev. 2004, 18, 1618–1629.
- De Magis, A.; Manzo, S.G.; Russo, M.; Marinello, J.; Morigi, R.; Sordet, O.; Capranico, G. DNA Damage and Genome Instability by G-Quadruplex Ligands Are Mediated by R Loops in Human Cancer Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 816–825.
- Yu, K.; Lieber, M.R. Current Insights into the Mechanism of Mammalian Immunoglobulin Class Switch Recombination. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 333–351.
- Stavnezer, J.; Schrader, C.E. Ig Heavy Chain Class Switch Recombination: Mechanism and Regulation. J. Immunol. 2014, 193, 5370–5378.
- Mathew, R.; Hartmuth, K.; Möhlmann, S.; Urlaub, H.; Ficner, R.; Lührmann, R. Phosphorylation of Human PRP28 by SRPK2 Is Required for Integration of the U4/U6-U5 Tri-SnRNP into the Spliceosome. Nat. Struct. Mol. Biol. 2008, 15, 435–443.
- Sridhara, S.C.; Carvalho, S.; Grosso, A.R.; Gallego-Paez, L.M.; Carmo-Fonseca, M.; de Almeida, S.F. Transcription Dynamics Prevent RNA-Mediated Genomic Instability through SRPK2-Dependent DDX23 Phosphorylation. Cell Rep. 2017, 18, 334–343.
- Fu, X.D. The Superfamily of Arginine/Serine-Rich Splicing Factors. RNA 1995, 1, 663–680.
- Mayeda, A.; Krainer, A.R. Regulation of Alternative Pre-MRNA Splicing by HnRNP A1 and Splicing Factor SF2. Cell 1992, 68, 365–375.
- Cáceres, J.F.; Kornblihtt, A.R. Alternative Splicing: Multiple Control Mechanisms and Involvement in Human Disease. Trends Genet. 2002, 18, 186–193.
- Li, X.; Manley, J.L. Inactivation of the SR Protein Splicing Factor ASF/SF2 Results in Genomic Instability. Cell 2005, 122, 365–378.
- Promonet, A.; Padioleau, I.; Liu, Y.; Sanz, L.; Biernacka, A.; Schmitz, A.-L.; Skrzypczak, M.; Sarrazin, A.; Mettling, C.; Rowicka, M.; et al. Topoisomerase 1 Prevents Replication Stress at R-Loop-Enriched Transcription Termination Sites. Nat. Commun. 2020, 11, 3940.
- Li, M.; Pokharel, S.; Wang, J.-T.; Xu, X.; Liu, Y. RECQ5-Dependent SUMOylation of DNA Topoisomerase I Prevents Transcription-Associated Genome Instability. Nat. Commun. 2015, 6, 6720.
- Wang, J.C. Cellular Roles of DNA Topoisomerases: A Molecular Perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440.
- Wu, H.Y.; Shyy, S.H.; Wang, J.C.; Liu, L.F. Transcription Generates Positively and Negatively Supercoiled Domains in the Template. Cell 1988, 53, 433–440.
- Marinello, J.; Bertoncini, S.; Aloisi, I.; Cristini, A.; Malagoli Tagliazucchi, G.; Forcato, M.; Sordet, O.; Capranico, G. Dynamic Effects of Topoisomerase I Inhibition on R-Loops and Short Transcripts at Active Promoters. PLoS ONE 2016, 11, e0147053.
- Obrdlik, A.; Kukalev, A.; Louvet, E.; Farrants, A.-K.O.; Caputo, L.; Percipalle, P. The Histone Acetyltransferase PCAF Associates with Actin and HnRNP U for RNA Polymerase II Transcription. Mol. Cell. Biol. 2008, 28, 6342–6357.
- Yugami, M.; Kabe, Y.; Yamaguchi, Y.; Wada, T.; Handa, H. HnRNP-U Enhances the Expression of Specific Genes by Stabilizing MRNA. FEBS Lett. 2007, 581, 1–7.
- Xiao, R.; Tang, P.; Yang, B.; Huang, J.; Zhou, Y.; Shao, C.; Li, H.; Sun, H.; Zhang, Y.; Fu, X.-D. Nuclear Matrix Factor HnRNP U/SAF-A Exerts a Global Control of Alternative Splicing by Regulating U2 SnRNP Maturation. Mol. Cell 2012, 45, 656–668.
- Britton, S.; Dernoncourt, E.; Delteil, C.; Froment, C.; Schiltz, O.; Salles, B.; Frit, P.; Calsou, P. DNA Damage Triggers SAF-A and RNA Biogenesis Factors Exclusion from Chromatin Coupled to R-Loops Removal. Nucleic Acids Res. 2014, 42, 9047–9062.
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817.
- Alfano, L.; Caporaso, A.; Altieri, A.; Dell’Aquila, M.; Landi, C.; Bini, L.; Pentimalli, F.; Giordano, A. Depletion of the RNA Binding Protein HNRNPD Impairs Homologous Recombination by Inhibiting DNA-End Resection and Inducing R-Loop Accumulation. Nucleic Acids Res. 2019, 47, 4068–4085.
- Yoon, J.-H.; De, S.; Srikantan, S.; Abdelmohsen, K.; Grammatikakis, I.; Kim, J.; Kim, K.M.; Noh, J.H.; White, E.J.F.; Martindale, J.L.; et al. PAR-CLIP Analysis Uncovers AUF1 Impact on Target RNA Fate and Genome Integrity. Nat. Commun. 2014, 5, 5248.
- Marchesini, M.; Ogoti, Y.; Fiorini, E.; Aktas Samur, A.; Nezi, L.; D’Anca, M.; Storti, P.; Samur, M.K.; Ganan-Gomez, I.; Fulciniti, M.T.; et al. ILF2 Is a Regulator of RNA Splicing and DNA Damage Response in 1q21-Amplified Multiple Myeloma. Cancer Cell 2017, 32, 88–100.e6.
- Okamoto, Y.; Abe, M.; Itaya, A.; Tomida, J.; Ishiai, M.; Takaori-Kondo, A.; Taoka, M.; Isobe, T.; Takata, M. FANCD2 Protects Genome Stability by Recruiting RNA Processing Enzymes to Resolve R-Loops during Mild Replication Stress. FEBS J. 2019, 286, 139–150.
- Jimeno, S.; Rondón, A.G.; Luna, R.; Aguilera, A. The Yeast THO Complex and MRNA Export Factors Link RNA Metabolism with Transcription and Genome Instability. EMBO J. 2002, 21, 3526–3535.
- Strässer, K.; Masuda, S.; Mason, P.; Pfannstiel, J.; Oppizzi, M.; Rodriguez-Navarro, S.; Rondón, A.G.; Aguilera, A.; Struhl, K.; Reed, R.; et al. TREX Is a Conserved Complex Coupling Transcription with Messenger RNA Export. Nature 2002, 417, 304–308.
- Luna, R.; Rondón, A.G.; Pérez-Calero, C.; Salas-Armenteros, I.; Aguilera, A. The THO Complex as a Paradigm for the Prevention of Cotranscriptional R-Loops. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2019; Volume 84, pp. 105–114.
- Gómez-González, B.; Felipe-Abrio, I.; Aguilera, A. The S-Phase Checkpoint Is Required To Respond to R-Loops Accumulated in THO Mutants. Mol. Cell. Biol. 2009, 29, 5203–5213.
- Gómez-González, B.; García-Rubio, M.; Bermejo, R.; Gaillard, H.; Shirahige, K.; Marín, A.; Foiani, M.; Aguilera, A. Genome-Wide Function of THO/TREX in Active Genes Prevents R-Loop-Dependent Replication Obstacles. EMBO J. 2011, 30, 3106–3119.
- Domínguez-Sánchez, M.S.; Barroso, S.; Gómez-González, B.; Luna, R.; Aguilera, A. Genome Instability and Transcription Elongation Impairment in Human Cells Depleted of THO/TREX. PLoS Genet. 2011, 7.
- Pérez-Calero, C.; Bayona-Feliu, A.; Xue, X.; Barroso, S.I.; Muñoz, S.; González-Basallote, V.M.; Sung, P.; Aguilera, A. UAP56/DDX39B Is a Major Cotranscriptional RNA–DNA Helicase That Unwinds Harmful R Loops Genome-Wide. Genes Dev. 2020.
- Salas-Armenteros, I.; Pérez-Calero, C.; Bayona-Feliu, A.; Tumini, E.; Luna, R.; Aguilera, A. Human THO–Sin3A Interaction Reveals New Mechanisms to Prevent R-loops That Cause Genome Instability. EMBO J. 2017, 36, 3532–3547.
- Cai, S.; Bai, Y.; Wang, H.; Zhao, Z.; Ding, X.; Zhang, H.; Zhang, X.; Liu, Y.; Jia, Y.; Li, Y.; et al. Knockdown of THOC1 Reduces the Proliferation of Hepatocellular Carcinoma and Increases the Sensitivity to Cisplatin. J. Exp. Clin. Cancer Res. 2020, 39, 135.
- Faza, M.B.; Kemmler, S.; Jimeno, S.; González-Aguilera, C.; Aguilera, A.; Hurt, E.; Panse, V.G. Sem1 Is a Functional Component of the Nuclear Pore Complex-Associated Messenger RNA Export Machinery. J. Cell Biol. 2009, 184, 833–846.
- Wilmes, G.M.; Bergkessel, M.; Bandyopadhyay, S.; Shales, M.; Braberg, H.; Cagney, G.; Collins, S.R.; Whitworth, G.B.; Kress, T.L.; Weissman, J.S.; et al. A Genetic Interaction Map of RNA-Processing Factors Reveals Links between Sem1/Dss1-Containing Complexes and MRNA Export and Splicing. Mol. Cell 2008, 32, 735–746.
- Gallardo, M.; Luna, R.; Erdjument-Bromage, H.; Tempst, P.; Aguilera, A. Nab2p and the Thp1p-Sac3p Complex Functionally Interact at the Interface between Transcription and MRNA Metabolism. J. Biol. Chem. 2003, 278, 24225–24232.
- Santos-Pereira, J.M.; García-Rubio, M.L.; González-Aguilera, C.; Luna, R.; Aguilera, A. A Genome-Wide Function of THSC/TREX-2 at Active Genes Prevents Transcription–Replication Collisions. Nucleic Acids Res. 2014, 42, 12000–12014.
- Bhatia, V.; Barroso, S.I.; García-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 Prevents R-Loop Accumulation and Associates with TREX-2 MRNA Export Factor PCID2. Nature 2014, 511, 362–365.
- Eberle, A.B.; Visa, N. Quality Control of MRNP Biogenesis: Networking at the Transcription Site. In Seminars in Cell and Developmental Biology; Academic Press: New York, NY, USA, 2014; Volume 32, pp. 37–46.
- Schmid, M.; Jensen, T.H. The Exosome: A Multipurpose RNA-Decay Machine. Trends Biochem. Sci. 2008, 33, 501–510.
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.-P.; Ahmann, G.J.; Adli, M.; et al. Initial Genome Sequencing and Analysis of Multiple Myeloma. Nature 2011, 471, 467–472.
- Boyle, E.M.; Ashby, C.; Tytarenko, R.G.; Deshpande, S.; Wang, H.; Wang, Y.; Rosenthal, A.; Sawyer, J.; Tian, E.; Flynt, E.; et al. BRAF and DIS3 Mutations Associate with Adverse Outcome in a Long-Term Follow-up of Patients with Multiple Myeloma. Clin. Cancer Res. 2020, 26, 2422–2432.
- Todoerti, K.; Ronchetti, D.; Favasuli, V.; Maura, F.; Morabito, F.; Bolli, N.; Taiana, E.; Neri, A. DIS3 Mutations in Multiple Myeloma Impact the Transcriptional Signature and Clinical Outcome. Arch. Ist. Ric. 2021.
- Pertesi, M.; Vallée, M.; Wei, X.; Revuelta, M.V.; Galia, P.; Demangel, D.; Oliver, J.; Foll, M.; Chen, S.; Perrial, E.; et al. Exome Sequencing Identifies Germline Variants in DIS3 in Familial Multiple Myeloma. Leukemia 2019, 33, 2324–2330.
- Domingo-Prim, J.; Endara-Coll, M.; Bonath, F.; Jimeno, S.; Prados-Carvajal, R.; Friedländer, M.R.; Huertas, P.; Visa, N. EXOSC10 Is Required for RPA Assembly and Controlled DNA End Resection at DNA Double-Strand Breaks. Nat. Commun. 2019, 10, 2135.
- Richard, P.; Feng, S.; Manley, J.L. A SUMO-Dependent Interaction between Senataxin and the Exosome, Disrupted in the Neurodegenerative Disease AOA2, Targets the Exosome to Sites of Transcription-Induced DNA Damage. Genes Dev. 2013, 27, 2227–2232.
- Ogami, K.; Chen, Y.; Manley, J.L. RNA Surveillance by the Nuclear RNA Exosome: Mechanisms and Significance. Noncoding RNA 2018, 4, 8.
- Kazadi, D.; Lim, J.; Rothschild, G.; Grinstein, V.; Laffleur, B.; Becherel, O.; Lavin, M.J.; Basu, U. Effects of Senataxin and RNA Exosome on B-Cell Chromosomal Integrity. Heliyon 2020, 6.
- Basu, U.; Meng, F.-L.; Keim, C.; Grinstein, V.; Pefanis, E.; Eccleston, J.; Zhang, T.; Myers, D.; Wasserman, C.R.; Wesemann, D.R.; et al. The RNA Exosome Targets the AID Cytidine Deaminase to Both Strands of Transcribed Duplex DNA Substrates. Cell 2011, 144, 353–363.
- Laffleur, B.; Lim, J.; Zhang, W.; Chen, Y.; Pefanis, E.; Bizarro, J.; Batista, C.R.; Wu, L.; Economides, A.N.; Wang, J.; et al. Noncoding RNA Processing by DIS3 Regulates Chromosomal Architecture and Somatic Hypermutation in B Cells. Nat. Genet. 2021, 53, 230–242.
- Kim, M.; Vasiljeva, L.; Rando, O.J.; Zhelkovsky, A.; Moore, C.; Buratowski, S. Distinct Pathways for SnoRNA and MRNA Termination. Mol. Cell 2006, 24, 723–734.
- West, S.; Gromak, N.; Proudfoot, N.J. Human 5′→3′ Exonuclease Xrn2 Promotes Transcription Termination at Co-Transcriptional Cleavage Sites. Nature 2004, 432, 522–525.
- Morales, J.C.; Richard, P.; Patidar, P.L.; Motea, E.A.; Dang, T.T.; Manley, J.L.; Boothman, D.A. XRN2 Links Transcription Termination to DNA Damage and Replication Stress. PLoS Genet. 2016, 12.
- Dang, T.T.; Morales, J.C. XRN2 Links RNA:DNA Hybrid Resolution to Double Strand Break Repair Pathway Choice. Cancers 2020, 12, 1821.
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human Senataxin Resolves RNA/DNA Hybrids Formed at Transcriptional Pause Sites to Promote Xrn2-Dependent Termination. Mol. Cell 2011, 42, 794–805.
- Hirling, H.; Scheffner, M.; Restle, T.; Stahl, H. RNA Helicase Activity Associated with the Human P68 Protein. Nature 1989, 339, 562–564.
- Xing, Z.; Wang, S.; Tran, E.J. Characterization of the Mammalian DEAD-Box Protein DDX5 Reveals Functional Conservation with S. Cerevisiae Ortholog Dbp2 in Transcriptional Control and Glucose Metabolism. RNA 2017, 23, 1125–1138.
- Mersaoui, S.Y.; Yu, Z.; Coulombe, Y.; Karam, M.; Busatto, F.F.; Masson, J.; Richard, S. Arginine Methylation of the DDX5 Helicase RGG/RG Motif by PRMT5 Regulates Resolution of RNA:DNA Hybrids. EMBO J. 2019, 38.
- Villarreal, O.D.; Mersaoui, S.Y.; Yu, Z.; Masson, J.-Y.; Richard, S. Genome-Wide R-Loop Analysis Defines Unique Roles for DDX5, XRN2, and PRMT5 in DNA/RNA Hybrid Resolution. Life Sci. Alliance 2020, 3.
- Gullà, A.; Hideshima, T.; Bianchi, G.; Fulciniti, M.; Kemal Samur, M.; Qi, J.; Tai, Y.-T.; Harada, T.; Morelli, E.; Amodio, N.; et al. Protein Arginine Methyltransferase 5 Has Prognostic Relevance and Is a Druggable Target in Multiple Myeloma. Leukemia 2018, 32, 996–1002.
- Bogliolo, M.; Surrallés, J. Fanconi Anemia: A Model Disease for Studies on Human Genetics and Advanced Therapeutics. Curr. Opin. Genet. Dev. 2015, 33, 32–40.
- Tischkowitz, M.D.; Hodgson, S.V. Fanconi Anaemia. J. Med. Genet. 2003, 40, 1–10.
- Alter, B.P.; Greene, M.H.; Velazquez, I.; Rosenberg, P.S. Cancer in Fanconi Anemia. Blood 2003, 101, 2072.
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi Anaemia Pathway: New Players and New Functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349.
- Mamrak, N.E.; Shimamura, A.; Howlett, N.G. Recent Discoveries in the Molecular Pathogenesis of the Inherited Bone Marrow Failure Syndrome Fanconi Anemia. Blood Rev. 2017, 31, 93–99.
- García-Rubio, M.L.; Pérez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi Anemia Pathway Protects Genome Integrity from R-Loops. PLoS Genet. 2015, 11, e1005674.
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.-C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cell 2015, 60, 351–361.
- Han, S.-S.; Tompkins, V.S.; Son, D.-J.; Han, S.; Yun, H.; Kamberos, N.L.; Dehoedt, C.L.; Gu, C.; Holman, C.; Tricot, G.; et al. CDKN1A and FANCD2 Are Potential Oncotargets in Burkitt Lymphoma and Multiple Myeloma. Exp. Hematol. Oncol. 2015, 4, 9.
- Madireddy, A.; Kosiyatrakul, S.T.; Gerhardt, J.; Boisvert, R.A.; Vuono, E.A.; Moyano, E.H.; Garcia Rubio, M.L.; Owen, N.; Yan, Z.; Olson, S.; et al. FANCD2 Facilitates Replication through Common Fragile Sites. Mol. Cell 2016, 64, 388–404.
- Liang, Z.; Liang, F.; Teng, Y.; Chen, X.; Liu, J.; Longerich, S.; Rao, T.; Green, A.M.; Collins, N.B.; Xiong, Y.; et al. Binding of FANCI-FANCD2 Complex to RNA and R-Loops Stimulates Robust FANCD2 Monoubiquitination. Cell Rep. 2019, 26, 564–572.e5.
- Liu, X.-P.; Yin, X.-H.; Meng, X.-Y.; Yan, X.-H.; Wang, F.; He, L. Development and Validation of a 9-Gene Prognostic Signature in Patients with Multiple Myeloma. Front. Oncol. 2019, 8, 615.
- Bohl, S.R.; Schmalbrock, L.K.; Bauhuf, I.; Meyer, T.; Dolnik, A.; Szyska, M.; Blätte, T.J.; Knödler, S.; Röhner, L.; Miller, D.; et al. Comprehensive CRISPR-Cas9 Screens Identify Genetic Determinants of Drug Responsiveness in Multiple Myeloma. Blood Adv. 2021, 5, 2391–2402.
- Chang, E.Y.-C.; Tsai, S.; Aristizabal, M.J.; Wells, J.P.; Coulombe, Y.; Busatto, F.F.; Chan, Y.A.; Kumar, A.; Dan Zhu, Y.; Wang, A.Y.-H.; et al. MRE11-RAD50-NBS1 Promotes Fanconi Anemia R-Loop Suppression at Transcription–Replication Conflicts. Nat. Commun. 2019, 10.
- Wu, Y.; Shin-ya, K.; Brosh, R.M. FANCJ Helicase Defective in Fanconia Anemia and Breast Cancer Unwinds G-Quadruplex DNA To Defend Genomic Stability. Mol. Cell. Biol. 2008, 28, 4116–4128.
- London, T.B.C.; Barber, L.J.; Mosedale, G.; Kelly, G.P.; Balasubramanian, S.; Hickson, I.D.; Boulton, S.J.; Hiom, K. FANCJ Is a Structure-Specific DNA Helicase Associated with the Maintenance of Genomic G/C Tracts. J. Biol. Chem. 2008, 283, 36132–36139.
- Wu, C.G.; Spies, M. G-Quadruplex Recognition and Remodeling by the FANCJ Helicase. Nucleic Acids Res. 2016, 44, 8742–8753.
- Castillo Bosch, P.; Segura-Bayona, S.; Koole, W.; van Heteren, J.T.; Dewar, J.M.; Tijsterman, M.; Knipscheer, P. FANCJ Promotes DNA Synthesis through G-Quadruplex Structures. EMBO J. 2014, 33, 2521–2533.
- Cantor, S.; Drapkin, R.; Zhang, F.; Lin, Y.; Han, J.; Pamidi, S.; Livingston, D.M. The BRCA1-Associated Protein BACH1 Is a DNA Helicase Targeted by Clinically Relevant Inactivating Mutations. Proc. Natl. Acad. Sci. USA 2004, 101, 2357–2362.
- Odermatt, D.C.; Lee, W.T.C.; Wild, S.; Jozwiakowski, S.K.; Rothenberg, E.; Gari, K. Cancer-Associated Mutations in the Iron-Sulfur Domain of FANCJ Affect G-Quadruplex Metabolism. PLoS Genet. 2020, 16.
- Lowran, K.; Campbell, L.; Popp, P.; Wu, C.G. Assembly of a G-Quadruplex Repair Complex by the FANCJ DNA Helicase and the REV1 Polymerase. Genes 2019, 11, 5.
- Eddy, S.; Ketkar, A.; Zafar, M.K.; Maddukuri, L.; Choi, J.-Y.; Eoff, R.L. Human Rev1 Polymerase Disrupts G-Quadruplex DNA. Nucleic Acids Res. 2014, 42, 3272–3285.
- Ketkar, A.; Smith, L.; Johnson, C.; Richey, A.; Berry, M.; Hartman, J.H.; Maddukuri, L.; Reed, M.R.; Gunderson, J.E.C.; Leung, J.W.C.; et al. Human Rev1 Relies on Insert-2 to Promote Selective Binding and Accurate Replication of Stabilized G-Quadruplex Motifs. Nucleic Acids Res. 2021, 49, 2065–2084.
- Nalepa, G.; Clapp, D.W. Fanconi Anaemia and Cancer: An Intricate Relationship. Nat. Rev. Cancer 2018, 18, 168–185.
- Savage, K.I.; Matchett, K.B.; Barros, E.M.; Cooper, K.M.; Irwin, G.W.; Gorski, J.J.; Orr, K.S.; Vohhodina, J.; Kavanagh, J.N.; Madden, A.F.; et al. BRCA1 Deficiency Exacerbates Estrogen-Induced DNA Damage and Genomic Instability. Cancer Res. 2014, 74, 2773–2784.
- Yarde, D.N.; Oliveira, V.; Mathews, L.; Wang, X.; Villagra, A.; Boulware, D.; Shain, K.H.; Hazlehurst, L.A.; Alsina, M.; Chen, D.-T.; et al. Targeting the Fanconi Anemia/BRCA Pathway Circumvents Drug Resistance in Multiple Myeloma. Cancer Res. 2009, 69, 9367–9375.
- Chen, Q.; Van der Sluis, P.C.; Boulware, D.; Hazlehurst, L.A.; Dalton, W.S. The FA/BRCA Pathway Is Involved in Melphalan-Induced DNA Interstrand Cross-Link Repair and Accounts for Melphalan Resistance in Multiple Myeloma Cells. Blood 2005, 106, 698–705.
- Volcic, M.; Karl, S.; Baumann, B.; Salles, D.; Daniel, P.; Fulda, S.; Wiesmüller, L. NF-ΚB Regulates DNA Double-Strand Break Repair in Conjunction with BRCA1–CtIP Complexes. Nucleic Acids Res. 2012, 40, 181–195.
- Demchenko, Y.N.; Kuehl, W.M. A Critical Role for the NFkB Pathway in Multiple Myeloma. Oncotarget 2010, 1, 59–68.
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 Recruitment to Transcriptional Pause Sites Is Required for R-Loop-Driven DNA Damage Repair. Mol. Cell 2015, 57, 636–647.
- Hill, S.J.; Rolland, T.; Adelmant, G.; Xia, X.; Owen, M.S.; Dricot, A.; Zack, T.I.; Sahni, N.; Jacob, Y.; Hao, T.; et al. Systematic Screening Reveals a Role for BRCA1 in the Response to Transcription-Associated DNA Damage. Genes Dev. 2014, 28, 1957–1975.
- Zhang, X.; Chiang, H.-C.; Wang, Y.; Zhang, C.; Smith, S.; Zhao, X.; Nair, S.J.; Michalek, J.; Jatoi, I.; Lautner, M.; et al. Attenuation of RNA Polymerase II Pausing Mitigates BRCA1-Associated R-Loop Accumulation and Tumorigenesis. Nat. Commun. 2017, 8, 15908.
- Sessa, G.; Gómez-González, B.; Silva, S.; Pérez-Calero, C.; Beaurepere, R.; Barroso, S.; Martineau, S.; Martin, C.; Ehlén, Å.; Martínez, J.S.; et al. BRCA2 Promotes R-Loop Resolution by DDX5 Helicase at DNA Breaks to Facilitate Their Repair by Homologous Recombination. EMBO J. 2021, 40, e106018.
- Yu, Z.; Mersaoui, S.Y.; Guitton-Sert, L.; Coulombe, Y.; Song, J.; Masson, J.-Y.; Richard, S. DDX5 Resolves R-Loops at DNA Double-Strand Breaks to Promote DNA Repair and Avoid Chromosomal Deletions. NAR Cancer 2020, 2.
- Mailand, N.; Gibbs-Seymour, I.; Bekker-Jensen, S. Regulation of PCNA-Protein Interactions for Genome Stability. Nat. Rev. Mol. Cell Biol. 2013, 14, 269–282.
- Alexandrakis, M.G.; Passam, F.H.; Pappa, C.A.; Dambaki, C.; Sfakiotaki, G.; Alegakis, A.K.; Kyriakou, D.S.; Stathopoulos, E. Expression of Proliferating Cell Nuclear Antigen (PCNA) in Multiple Myeloma: Its Relationship to Bone Marrow Microvessel Density and Other Factors of Disease Activity. Int. J. Immunopathol. Pharmacol. 2004, 17, 49–56.
- Tsirakis, G.; Pappa, C.A.; Kaparou, M.; Katsomitrou, V.; Hatzivasili, A.; Alegakis, T.; Xekalou, A.; Stathopoulos, E.N.; Alexandrakis, M.G. Assessment of Proliferating Cell Nuclear Antigen and Its Relationship with Proinflammatory Cytokines and Parameters of Disease Activity in Multiple Myeloma Patients. Eur. J. Histochem. 2011, 55, e21.
- Müller, R.; Misund, K.; Holien, T.; Bachke, S.; Gilljam, K.M.; Våtsveen, T.K.; Rø, T.B.; Bellacchio, E.; Sundan, A.; Otterlei, M. Targeting Proliferating Cell Nuclear Antigen and Its Protein Interactions Induces Apoptosis in Multiple Myeloma Cells. PLoS ONE 2013, 8, e70430.
- Moldovan, G.-L.; Dejsuphong, D.; Petalcorin, M.I.R.; Hofmann, K.; Takeda, S.; Boulton, S.J.; D’Andrea, A.D. Inhibition of Homologous Recombination by the PCNA-Interacting Protein PARI. Mol. Cell 2012, 45, 75–86.
- Gali, H.; Juhasz, S.; Morocz, M.; Hajdu, I.; Fatyol, K.; Szukacsov, V.; Burkovics, P.; Haracska, L. Role of SUMO Modification of Human PCNA at Stalled Replication Fork. Nucleic Acids Res. 2012, 40, 6049–6059.
- Li, M.; Xu, X.; Chang, C.-W.; Zheng, L.; Shen, B.; Liu, Y. SUMO2 Conjugation of PCNA Facilitates Chromatin Remodeling to Resolve Transcription-Replication Conflicts. Nat. Commun. 2018, 9.
- Xie, H.; Gu, Y.; Wang, W.; Wang, X.; Ye, X.; Xin, C.; Lu, M.; Reddy, B.A.; Shu, P. Silencing of SENP2 in Multiple Myeloma Induces Bortezomib Resistance by Activating NF-ΚB Through the Modulation of IκBα Sumoylation. Sci. Rep. 2020, 10, 766.
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP Promotes DNA End Resection. Nature 2007, 450, 509–514.
- Symington, L.S. Mechanism and Regulation of DNA End Resection in Eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212.
- Makharashvili, N.; Tubbs, A.T.; Yang, S.-H.; Wang, H.; Barton, O.; Zhou, Y.; Deshpande, R.A.; Lee, J.-H.; Lobrich, M.; Sleckman, B.P.; et al. Catalytic and Noncatalytic Roles of the CtIP Endonuclease in Double-Strand Break End Resection. Mol. Cell 2014, 54, 1022–1033.
- Zhang, W.; Song, Y.; He, X.; Liu, X.; Zhang, Y.; Yang, Z.; Yang, P.; Wang, J.; Hu, K.; Liu, W.; et al. Prognosis Value of RBBP8 Expression in Plasma Cell Myeloma. Cancer Gene Ther. 2020, 27, 22–29.
- Makharashvili, N.; Arora, S.; Yin, Y.; Fu, Q.; Wen, X.; Lee, J.-H.; Kao, C.-H.; Leung, J.W.; Miller, K.M.; Paull, T.T. Sae2/CtIP Prevents R-Loop Accumulation in Eukaryotic Cells. eLife 2018, 7.
- Keijzers, G.; Liu, D.; Rasmussen, L.J. Exonuclease 1 and Its Versatile Roles in DNA Repair. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 440–451.
- Stroik, S.; Kurtz, K.; Lin, K.; Karachenets, S.; Myers, C.L.; Bielinsky, A.-K.; Hendrickson, E.A. EXO1 Resection at G-Quadruplex Structures Facilitates Resolution and Replication. Nucleic Acids Res. 2020.
- Dehé, P.-M.; Gaillard, P.-H.L. Control of Structure-Specific Endonucleases to Maintain Genome Stability. Nat. Rev. Mol. Cell Biol. 2017, 18, 315–330.
- Tian, M.; Alt, F.W. Transcription-Induced Cleavage of Immunoglobulin Switch Regions by Nucleotide Excision Repair Nucleases in vitro. J. Biol. Chem. 2000, 275, 24163–24172.
- Sollier, J.; Stork, C.T.; García-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-Coupled Nucleotide Excision Repair Factors Promote R-Loop-Induced Genome Instability. Mol. Cell 2014, 56, 777–785.
- Cristini, A.; Ricci, G.; Britton, S.; Salimbeni, S.; Huang, S.N.; Marinello, J.; Calsou, P.; Pommier, Y.; Favre, G.; Capranico, G.; et al. Dual Processing of R-Loops and Topoisomerase I Induces Transcription-Dependent DNA Double-Strand Breaks. Cell Rep. 2019, 28, 3167–3181.
- Yasuhara, T.; Kato, R.; Hagiwara, Y.; Shiotani, B.; Yamauchi, M.; Nakada, S.; Shibata, A.; Miyagawa, K. Human Rad52 Promotes XPG-Mediated R-Loop Processing to Initiate Transcription-Associated Homologous Recombination Repair. Cell 2018, 175, 558–570.e11.
- Lin, Y.-L.; Pasero, P. Caught in the Act: R-Loops Are Cleaved by Structure-Specific Endonucleases to Generate DSBs. Mol. Cell 2014, 56, 721–722.
- de Larrea, C.F.; Navarro, A.; Tovar, N.; Pedrosa, F.; Díaz, T.; Cibeira, M.T.; Magnano, L.; Rosiñol, L.; Rovira, M.; Rozman, M.; et al. Impact of Single Nucleotide Polymorphisms in Genes Involved in DNA Repair and Drug Metabolism On Survival After Autologous Stem Cell Transplantation in Patients with Multiple Myeloma. Blood 2012, 120, 2934.
- He, Y.; Pasupala, N.; Zhi, H.; Dorjbal, B.; Hussain, I.; Shih, H.-M.; Bhattacharyya, S.; Biswas, R.; Miljkovic, M.; Semmes, O.J.; et al. NF-ΚB–Induced R-Loop Accumulation and DNA Damage Select for Nucleotide Excision Repair Deficiencies in Adult T Cell Leukemia. Proc. Natl. Acad. Sci. USA 2021, 118.
- Ellis, N.A.; Groden, J.; Ye, T.Z.; Straughen, J.; Lennon, D.J.; Ciocci, S.; Proytcheva, M.; German, J. The Bloom’s Syndrome Gene Product Is Homologous to RecQ Helicases. Cell 1995, 83, 655–666.
- Sun, H.; Karow, J.K.; Hickson, I.D.; Maizels, N. The Bloom’s Syndrome Helicase Unwinds G4 DNA. J. Biol. Chem. 1998, 273, 27587–27592.
- Popuri, V.; Bachrati, C.Z.; Muzzolini, L.; Mosedale, G.; Costantini, S.; Giacomini, E.; Hickson, I.D.; Vindigni, A. The Human RecQ Helicases, BLM and RECQ1, Display Distinct DNA Substrate Specificities. J. Biol. Chem. 2008, 283, 17766–17776.
- Wu, W.-Q.; Hou, X.-M.; Li, M.; Dou, S.-X.; Xi, X.-G. BLM Unfolds G-Quadruplexes in Different Structural Environments through Different Mechanisms. Nucleic Acids Res. 2015, 43, 4614–4626.
- Chang, E.Y.-C.; Novoa, C.A.; Aristizabal, M.J.; Coulombe, Y.; Segovia, R.; Chaturvedi, R.; Shen, Y.; Keong, C.; Tam, A.S.; Jones, S.J.M.; et al. RECQ-like Helicases Sgs1 and BLM Regulate R-Loop–Associated Genome Instability. J. Cell Biol. 2017, 216, 3991–4005.
- van Wietmarschen, N.; Merzouk, S.; Halsema, N.; Spierings, D.C.J.; Guryev, V.; Lansdorp, P.M. BLM Helicase Suppresses Recombination at G-Quadruplex Motifs in Transcribed Genes. Nat. Commun. 2018, 9.
- Nguyen, G.H.; Tang, W.; Robles, A.I.; Beyer, R.P.; Gray, L.T.; Welsh, J.A.; Schetter, A.J.; Kumamoto, K.; Wang, X.W.; Hickson, I.D.; et al. Regulation of Gene Expression by the BLM Helicase Correlates with the Presence of G-Quadruplex DNA Motifs. Proc. Natl. Acad. Sci. USA 2014, 111, 9905–9910.
- Huppert, J.L.; Balasubramanian, S. Prevalence of Quadruplexes in the Human Genome. Nucleic Acids Res. 2005, 33, 2908–2916.
- Wu, W.; Rokutanda, N.; Takeuchi, J.; Lai, Y.; Maruyama, R.; Togashi, Y.; Nishikawa, H.; Arai, N.; Miyoshi, Y.; Suzuki, N.; et al. HERC2 Facilitates BLM and WRN Helicase Complex Interaction with RPA to Suppress G-Quadruplex DNA. Cancer Res. 2018, 78, 6371–6385.
- Rodriguez, R.; Müller, S.; Yeoman, J.A.; Trentesaux, C.; Riou, J.-F.; Balasubramanian, S. A Novel Small Molecule That Alters Shelterin Integrity and Triggers a DNA-Damage Response at Telomeres. J. Am. Chem. Soc. 2008, 130, 15758–15759.
- Lai, Y.; Zhu, M.; Wu, W.; Rokutanda, N.; Togashi, Y.; Liang, W.; Ohta, T. HERC2 Regulates RPA2 by Mediating ATR-Induced Ser33 Phosphorylation and Ubiquitin-Dependent Degradation. Sci. Rep. 2019, 9.
- Grierson, P.M.; Acharya, S.; Groden, J. Collaborating Functions of BLM and DNA Topoisomerase I in Regulating Human RDNA Transcription. Mutat. Res. 2013, 743, 89–96.
- Kassambara, A.; Herviou, L.; Ovejero, S.; Jourdan, M.; Thibaut, C.; Vikova, V.; Pasero, P.; Elemento, O.; Moreaux, J. RNA-Sequencing Data-Driven Dissection of Human Plasma Cell Differentiation Reveals New Potential Transcription Regulators. Leukemia 2021.
- Popuri, V.; Croteau, D.L.; Brosh, R.M.; Bohr, V.A. RECQ1 Is Required for Cellular Resistance to Replication Stress and Catalyzes Strand Exchange on Stalled Replication Fork Structures. Cell Cycle 2012, 11, 4252–4265.
- Lu, X.; Parvathaneni, S.; Hara, T.; Lal, A.; Sharma, S. Replication Stress Induces Specific Enrichment of RECQ1 at Common Fragile Sites FRA3B and FRA16D. Mol. Cancer 2013, 12, 29.
- Cui, S.; Arosio, D.; Doherty, K.M.; Brosh, R.M.; Falaschi, A.; Vindigni, A. Analysis of the Unwinding Activity of the Dimeric RECQ1 Helicase in the Presence of Human Replication Protein A. Nucleic Acids Res. 2004, 32, 2158–2170.
- Viziteu, E.; Klein, B.; Basbous, J.; Lin, Y.-L.; Hirtz, C.; Gourzones, C.; Tiers, L.; Bruyer, A.; Vincent, L.; Grandmougin, C.; et al. RECQ1 Helicase Is Involved in Replication Stress Survival and Drug Resistance in Multiple Myeloma. Leukemia 2017, 31, 2104–2113.
- Parvathaneni, S.; Lu, X.; Chaudhary, R.; Lal, A.; Madhusudan, S.; Sharma, S. RECQ1 Expression Is Upregulated in Response to DNA Damage and in a P53-Dependent Manner. Oncotarget 2017, 8, 75924–75942.
- Li, X.L.; Lu, X.; Parvathaneni, S.; Bilke, S.; Zhang, H.; Thangavel, S.; Vindigni, A.; Hara, T.; Zhu, Y.; Meltzer, P.S.; et al. Identification of RECQ1-Regulated Transcriptome Uncovers a Role of RECQ1 in Regulation of Cancer Cell Migration and Invasion. Cell Cycle 2014, 13, 2431–2445.
- Lu, X.; Parvathaneni, S.; Li, X.L.; Lal, A.; Sharma, S. Transcriptome Guided Identification of Novel Functions of RECQ1 Helicase. Methods 2016, 108, 111–117.
- Debnath, S.; Sharma, S. RECQ1 Helicase in Genomic Stability and Cancer. Genes 2020, 11, 622.
- Edwards, A.D.; Marecki, J.C.; Byrd, A.K.; Gao, J.; Raney, K.D. G-Quadruplex Loops Regulate PARP-1 Enzymatic Activation. Nucleic Acids Res. 2020.
- Bochman, M.L.; Sabouri, N.; Zakian, V.A. Unwinding the Functions of the Pif1 Family Helicases. DNA Repair 2010, 9, 237–249.
- Byrd, A.K.; Raney, K.D. A Parallel Quadruplex DNA Is Bound Tightly but Unfolded Slowly by Pif1 Helicase. J. Biol. Chem. 2015, 290, 6482–6494.
- Hou, X.-M.; Wu, W.-Q.; Duan, X.-L.; Liu, N.-N.; Li, H.-H.; Fu, J.; Dou, S.-X.; Li, M.; Xi, X.-G. Molecular Mechanism of G-Quadruplex Unwinding Helicase: Sequential and Repetitive Unfolding of G-Quadruplex by Pif1 Helicase. Biochem. J. 2015, 466, 189–199.
- Zhou, R.; Zhang, J.; Bochman, M.L.; Zakian, V.A.; Ha, T. Periodic DNA Patrolling Underlies Diverse Functions of Pif1 on R-Loops and G-Rich DNA. eLife 2014, 3, e02190.
- Dahan, D.; Tsirkas, I.; Dovrat, D.; Sparks, M.A.; Singh, S.P.; Galletto, R.; Aharoni, A. Pif1 Is Essential for Efficient Replisome Progression through Lagging Strand G-Quadruplex DNA Secondary Structures. Nucleic Acids Res. 2018, 46, 11847–11857.
- Paeschke, K.; Capra, J.A.; Zakian, V.A. DNA Replication through G-Quadruplex Motifs Is Promoted by the Saccharomyces Cerevisiae Pif1 DNA Helicase. Cell 2011, 145, 678–691.
- Ribeyre, C.; Lopes, J.; Boulé, J.-B.; Piazza, A.; Guédin, A.; Zakian, V.A.; Mergny, J.-L.; Nicolas, A. The Yeast Pif1 Helicase Prevents Genomic Instability Caused by G-Quadruplex-Forming CEB1 Sequences In Vivo. PLoS Genet. 2009, 5.
- Sparks, M.A.; Singh, S.P.; Burgers, P.M.; Galletto, R. Complementary Roles of Pif1 Helicase and Single Stranded DNA Binding Proteins in Stimulating DNA Replication through G-Quadruplexes. Nucleic Acids Res. 2019, 47, 8595–8605.
- Maestroni, L.; Audry, J.; Luciano, P.; Coulon, S.; Géli, V.; Corda, Y. RPA and Pif1 Cooperate to Remove G-Rich Structures at Both Leading and Lagging Strand. Cell Stress 2020, 4, 48–63.
- Safa, L.; Gueddouda, N.M.; Thiébaut, F.; Delagoutte, E.; Petruseva, I.; Lavrik, O.; Mendoza, O.; Bourdoncle, A.; Alberti, P.; Riou, J.-F.; et al. 5′ to 3′ Unfolding Directionality of DNA Secondary Structures by Replication Protein A. J. Biol. Chem. 2016, 291, 21246–21256.
- Salas, T.R.; Petruseva, I.; Lavrik, O.; Bourdoncle, A.; Mergny, J.-L.; Favre, A.; Saintomé, C. Human Replication Protein A Unfolds Telomeric G-Quadruplexes. Nucleic Acids Res. 2006, 34, 4857–4865.
- Paeschke, K.; Bochman, M.L.; Garcia, P.D.; Cejka, P.; Friedman, K.L.; Kowalczykowski, S.C.; Zakian, V.A. Pif1 Family Helicases Suppress Genome Instability at G-Quadruplex Motifs. Nature 2013, 497, 458–462.
- Audry, J.; Maestroni, L.; Delagoutte, E.; Gauthier, T.; Nakamura, T.M.; Gachet, Y.; Saintomé, C.; Géli, V.; Coulon, S. RPA Prevents G-Rich Structure Formation at Lagging-Strand Telomeres to Allow Maintenance of Chromosome Ends. EMBO J. 2015, 34, 1942–1958.
- Chib, S.; Byrd, A.K.; Raney, K.D. Yeast Helicase Pif1 Unwinds RNA:DNA Hybrids with Higher Processivity than DNA:DNA Duplexes. J. Biol. Chem. 2016, 291, 5889–5901.
- Tran, P.L.T.; Pohl, T.J.; Chen, C.-F.; Chan, A.; Pott, S.; Zakian, V.A. PIF1 Family DNA Helicases Suppress R-Loop Mediated Genome Instability at TRNA Genes. Nat. Commun. 2017, 8.
- Budd, M.E.; Choe, W.C.; Campbell, J.L. DNA2 Encodes a DNA Helicase Essential for Replication of Eukaryotic Chromosomes. J. Biol. Chem. 1995, 270, 26766–26769.
- Budd, M.E.; Reis, C.C.; Smith, S.; Myung, K.; Campbell, J.L. Evidence Suggesting That Pif1 Helicase Functions in DNA Replication with the Dna2 Helicase/Nuclease and DNA Polymerase Delta. Mol. Cell. Biol. 2006, 26, 2490–2500.
- Masuda-Sasa, T.; Polaczek, P.; Peng, X.P.; Chen, L.; Campbell, J.L. Processing of G4 DNA by Dna2 Helicase/Nuclease and Replication Protein A (RPA) Provides Insights into the Mechanism of Dna2/RPA Substrate Recognition. J. Biol. Chem. 2008, 283, 24359–24373.
- Lin, W.; Sampathi, S.; Dai, H.; Liu, C.; Zhou, M.; Hu, J.; Huang, Q.; Campbell, J.; Shin-Ya, K.; Zheng, L.; et al. Mammalian DNA2 Helicase/Nuclease Cleaves G-Quadruplex DNA and Is Required for Telomere Integrity. EMBO J. 2013, 32, 1425–1439.
- Choe, W.; Budd, M.; Imamura, O.; Hoopes, L.; Campbell, J.L. Dynamic Localization of an Okazaki Fragment Processing Protein Suggests a Novel Role in Telomere Replication. Mol. Cell. Biol. 2002, 22, 4202–4217.
- Karanja, K.K.; Cox, S.W.; Duxin, J.P.; Stewart, S.A.; Campbell, J.L. DNA2 and EXO1 in Replication-Coupled, Homology-Directed Repair and in the Interplay between HDR and the FA/BRCA Network. Cell Cycle 2012, 11, 3983–3996.
- Peng, G.; Dai, H.; Zhang, W.; Hsieh, H.-J.; Pan, M.-R.; Park, Y.-Y.; Tsai, Y.-L.; Bedrosian, I.; Lee, J.-S.; Ira, G.; et al. Human Nuclease/Helicase DNA2 Alleviates Replication Stress by Promoting DNA End Resection. Cancer Res. 2012, 72, 2802–2813.
- Daley, J.M.; Chiba, T.; Xue, X.; Niu, H.; Sung, P. Multifaceted Role of the Topo IIIα-RMI1-RMI2 Complex and DNA2 in the BLM-Dependent Pathway of DNA Break End Resection. Nucleic Acids Res. 2014, 42, 11083–11091.
- Palm, W.; de Lange, T. How Shelterin Protects Mammalian Telomeres. Annu. Rev. Genet. 2008, 42, 301–334.
- Zaug, A.J.; Podell, E.R.; Cech, T.R. Human POT1 Disrupts Telomeric G-Quadruplexes Allowing Telomerase Extension in vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 10864–10869.
- Gomez, D.; O’Donohue, M.-F.; Wenner, T.; Douarre, C.; Macadré, J.; Koebel, P.; Giraud-Panis, M.-J.; Kaplan, H.; Kolkes, A.; Shin-ya, K.; et al. The G-Quadruplex Ligand Telomestatin Inhibits POT1 Binding to Telomeric Sequences in vitro and Induces GFP-POT1 Dissociation from Telomeres in Human Cells. Cancer Res. 2006, 66, 6908–6912.
- Di Antonio, M.; Biffi, G.; Mariani, A.; Raiber, E.-A.; Rodriguez, R.; Balasubramanian, S. Selective RNA versus DNA G-Quadruplex Targeting by in Situ Click Chemistry. Angew. Chem. 2012, 51, 11073–11078.
- Ray, S.; Bandaria, J.N.; Qureshi, M.H.; Yildiz, A.; Balci, H. G-Quadruplex Formation in Telomeres Enhances POT1/TPP1 Protection against RPA Binding. Proc. Natl. Acad. Sci. USA 2014, 111, 2990–2995.
- Chaires, J.B.; Gray, R.D.; Dean, W.L.; Monsen, R.; DeLeeuw, L.W.; Stribinskis, V.; Trent, J.O. Human POT1 Unfolds G-Quadruplexes by Conformational Selection. Nucleic Acids Res. 2020.
- Paeschke, K.; Simonsson, T.; Postberg, J.; Rhodes, D.; Lipps, H.J. Telomere End-Binding Proteins Control the Formation of G-Quadruplex DNA Structures in Vivo. Nat. Struct. Mol. Biol. 2005, 12, 847–854.
- Panero, J.; Stanganelli, C.; Arbelbide, J.; Fantl, D.B.; Kohan, D.; García Rivello, H.; Rabinovich, G.A.; Slavutsky, I. Expression Profile of Shelterin Components in Plasma Cell Disorders. Clinical Significance of POT1 Overexpression. Blood Cells Mol. Dis. 2014, 52, 134–139.
- Venturutti, L.; Melnick, A.M. The Dangers of Déjà vu: Memory B Cells as the Cells of Origin of ABC-DLBCLs. Blood 2020, 136, 2263–2274.
- Rustad, E.H.; Yellapantula, V.; Leongamornlert, D.; Bolli, N.; Ledergor, G.; Nadeu, F.; Angelopoulos, N.; Dawson, K.J.; Mitchell, T.J.; Osborne, R.J.; et al. Timing the Initiation of Multiple Myeloma. Nat. Commun. 2020, 11.
More
Information
Subjects:
Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
810
Revisions:
2 times
(View History)
Update Date:
26 Sep 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No