 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jian Yi Gerald Goie | + 6355 word(s) | 6355 | 2020-07-22 06:17:55 | | | |
| 2 | Rita Xu | -3575 word(s) | 2780 | 2020-07-28 05:39:28 | | | | |
| 3 | Rita Xu | -46 word(s) | 2734 | 2020-10-26 09:45:12 | | |
Video Upload Options
Influenza A viral (IAV) infections are common, yet persistent as one of the major contributors towards respiratory viral diseases. With the complete eradication of IAVs seemingly impossible, IAV infections are of major public health concern globally as evident by the need for constant surveillance and vaccine renewals. This entry focuses on the innate immune response against influenza infections and in particular, the roles of mitogen-activated protein kinases (MAPKs) in this response. We first detailed the conventional methods of pathogen recognition of influenza viruses by pathogen recognition receptors (PRRs), leading to the activation of pathways involved in the anti-viral response. Predominantly, we have highlighted the roles that MAPKs (ERK, p38 and JNK) play in the activation of Type I Interferons (IFNs) and pro-inflammatory cytokines to resolve IAV infections. Taking a step further, we also looked at how highly pathogenic influenza A viruses (HPIAVs), as well as aberrant and dysfunctional signalling of the MAPK pathways may lead to a hyperactive immune response that is unwarranted, leading to the progression into acute lung injuries and acute respiratory distress syndrome (ARDS) from a simple infection. Taken together, we hope that this entry may shed some light on the important roles that MAPKs play in the innate immune response towards IAV infections, and to provide important considerations when tackling this global challenge.
1. Definition
Influenza is a major respiratory viral disease caused by infections from the influenza A virus (IAV) that persists across various seasonal outbreaks globally each year. Host immune response is a key factor determining disease severity of influenza infection, presenting an attractive target for the development of novel therapies for treatments. Among the multiple signal transduction pathways regulating the host immune activation and function in response to IAV infections, the mitogen-activated protein kinase (MAPK) pathways are important signalling axes, downstream of various pattern recognition receptors (PRRs), activated by IAVs that regulate various cellular processes in immune cells of both innate and adaptive immunity. Moreover, aberrant MAPK activation underpins overexuberant production of inflammatory mediators, promoting the development of the “cytokine storm”, a characteristic of severe respiratory viral diseases. Therefore, elucidation of the regulatory roles of MAPK in immune responses against IAVs is not only essential for understanding the pathogenesis of severe influenza, but also critical for developing MAPK-dependent therapies for treatment of respiratory viral diseases.
2. Introduction
Viruses are major contributors of infectious diseases, causing significant morbidity and mortality worldwide [1]. Unlike bacteria, the life cycle of viruses involves host cell entries, viral genome replications, viral protein productions, assemblies of viral proteins and, lastly, the release from host cells [2][3]. As such, viruses are greatly dependent on host’s cellular machinery for virulence and survival. Fortunately, both innate and adaptive immunities have evolved and developed various ways to detect and eliminate virus and infected cells in response to viral infections. Surface viral proteins, such as the capsid and viral genomes, act as pathogen-associated molecular patterns (PAMPs), which are recognised by pathogen recognition receptors (PRRs) on immune cells [4]. They commonly include Toll-like receptors (TLRs), Retinoic Acid-Inducible Gene-I (RIG-I)-like receptors (RLRs) and Nod-like receptors (NLRs) that recognise both extracellular and intracellular viral-associated molecular patterns [5]. Upon recognition, adaptor proteins like MyD88 are recruited to activate downstream signalling, through a phosphorylation cascade, that leads to the activation of pro-inflammatory cytokines and Type I Interferons (IFNs) to induce an anti-viral state [5]. Likewise, in response to the selection pressure from the host immune response, the rapid yet error-prone replication of viruses allows them to constantly generate mutants that aid in the adaptation and evasion of host immunity [6]. This is clearly demonstrated in the never-ending persistence of global infections caused by the influenza A virus (IAV) every year due to its high rates of mutation [7].
3. MAPKs and Innate Immunity to IAVs
The innate immunity is important for viral containment, and if possible, elimination, upon infection and plays a pivotal role in the subsequent induction and regulation of the adaptive immunity. When left unchecked, an excessive innate immune response contributes greatly to the development of respiratory pathologies such as ARDS [8][9]. Over the past few decades, considerable amount of research efforts have been devoted to uncover the mechanisms underlying the recognition and containment of IAVs by the innate immunity [10][11]. Of which, a number of studies have investigated the roles of virus-sensing receptors, generation of antiviral effector cells and the underlying molecular mechanisms related to MAPKs, which demonstrated the importance of MAPKs in innate recognition, activation of the IFN system, and expression of cytokines and chemokines in response to IAV antigens.
3.1. Innate Recognition of IAV Infection
Innate immunity against IAV infections is induced upon recognition of viral ssRNA by various PRRs at various subcellular locations during its replication process. The replication cycle of IAVs begins from the entry of host cells through HA-mediated endocytosis [12]. The acidic environment within the endosome promotes conformational changes of the HA glycoprotein, exposing fusion peptides that facilitate pore formation on the endosomal membranes which allows for the viral genome to be released into the cytoplasm. Subsequently, nuclear translocation occurs where the viral RNAs are transcribed and replicated for the synthesis of new viral genomes before nuclear export for translation of proteins using the hosts’ cytoplasmic and endoplasmic reticulum (ER)-associated ribosomes [12]. During this process, the viral genome is recognized by the host’s PRRs: RLRs in the cytoplasm, TLR3 and TLR7 in the endosome [13][14]. Activation of the PRRs by IAVs in the epithelial and immune cells residing in the respiratory tract results in the activation of the IRF3, NFκB and MAPK signal transduction pathways. This in turn regulates the expression of the Type I IFNs (IFNα and IFNβ), pro-inflammatory cytokines such as interleukin 6 (IL-6) and tumour necrotic factor α (TNFα), as well as chemokines like monocyte chemoattractant protein-1 (MCP-1) [15]. For instance, within the cytosol of infected cells, the 5′-triphosphate ends of the viral ssRNA are recognized by RIG-I, which results in its activation and translocation to the mitochondria to activate the mitochondrial antiviral signalling protein (MAVS) for downstream signalling [16][17][18][19]. On the other hand, viral genome in endosomes are detected by TLRs including TLR7, which transduces signals through the MyD88 adaptor protein to activate IRF7, MAPKs and NFκB for the expression of Type I IFNs and other pro-inflammatory mediators [19][20]. Interestingly, some existing evidence suggests that RLR and TLRs may not necessarily be indispensable in the activation of immune response against IAV infections. A recent study by Wu et al. showed that deficiency in RLR did not affect the survivability of C57BL/6 mice after lethal influenza infections [21]. An earlier study by Jeisy-Scott et al. also showed that the loss of TLR7 did not impact memory CD8+ T-cell response, and only moderately affected the development of B-cell adaptive immune memory [22]. IAV infections may also activate the NLRP3 inflammasome pathway [23][24]. ssRNAs of IAVs were shown to activate the NLRP3 inflammasome, leading to a lysosomal maturation and reactive oxygen species (ROS)-dependent production of IL-1β and IL-18 [25]. NLRP3 inflammasome may also be activated through the proton-specific Matrix-2 (M2) ion channel encoded by IAVs, which plays a role in the acidification of the virus-containing endosomes leading to membrane fusion and release of virions into the cytosol [26]. M2 localises to the Golgi upon infection, leading to the acidification of the Golgi compartment, which activates NLRP3 inflammasome and IL-1β production. Lastly, the virulence factor PB1-F2 of IAVs is also able to induce the production of IL-1β through a NLRP3- and Caspase-1-dependent pathway [27].
3.2. MAPKs and Expression of Inflammatory Mediators in Response to IAVs
Once influenza viruses are recognized by PRRs, inflammatory mediators, including cytokines, chemokines and other antimicrobial factors, are secreted by various types of cells, including epithelial cells, endothelial cells and monocytes/macrophages. Among the cells that are capable of secreting cytokines/chemokines, airway epithelial cells and tissue-resident alveolar macrophages are the major and immediate sources of inflammatory mediators in response to respiratory tissue assaults [28][29][30][31]. Apart from the induction of Type I IFN response, airway epithelial cells also secrete a number of cytokines, including IL-6, TNFα, granulocyte colony stimulating factor (G-CSF) and granulocyte macrophage colony stimulating factor (GM-CSF), which are all essential for anti-influenza immunity [32][33]. For instance, IL-6 drives the transition from innate to adaptive immunity, whereas TNFα amplifies cytotoxic activity and impairs viral replication [34][35]. Likewise, G-CSF and GM-CSF are required for the differentiation of myeloid cells, such as alveolar monocytes/macrophages in particular, promoting their effector functions and reducing influenza-mediated morbidity and mortality [36][37][38].
Chemokines secreted by airway epithelial cells and pulmonary macrophages recruit both innate and adaptive immune cells to the lung to amplify the immune response and release of cytotoxic and inflammatory factors [32]. CXCL8 recruits neutrophils to the lungs, whereas IP-10/CXCL10 and RANTES/CCL5 promotes the infiltration of monocytes, NK cells and T-cells from the peripheral circulation into the lungs [39][40][41][42]. Once recruited, these cells work specifically and cooperatively to control and eradicate the viruses from the airways and lungs. Neutrophils are the first few immune cells recruited to the site of infection, playing a crucial role in clearing virions or dead viral-infected cellular bodies [43][44]. As their effector functions hinge on the use of antimicrobial peptides and proteolytic granules, influenza viral particles can be quickly degraded and cleared. Alveolar macrophages on the other hand, are the major source of Type I IFNs which are essential for recruiting inflammatory monocytes to the lungs and generating an antiviral state during early viral infections [45][46]. Upon secretion, Type I IFNs bind to their respective receptors on neighbouring uninfected cells which, stimulating the expression of ISGs which ultimately induce a cell-intrinsic antiviral state, responsible for effective inhibition of IAV spreading and infection through various mechanisms [47]. The expression of Type I IFNs may be regulated by MAPKs through the regulation of IRF3 or direct transcription of IFN genes. It is known that JNK regulates IFNβ expression through the activating protein-1 (AP-1) transcription factor in response to IAV infections [48]. ERK, JNK and p38 were also shown to increase the expression of heme oxygenase-1 (HO-1) through the induction of nuclear factor erythroid 2-related factor 2 (Nrf2), thereby promoting the expression of Type I IFNs to suppress IAV infections [49][50]. However, further studies are required to further establish such novel mechanisms of MAPKs in innate immunity against IAVs.
In addition to the expression and action of Type I IFNs, activation of the MAPKs in these cells during IAV infection also regulate the expression of inflammatory mediators. Indeed, in human bronchial epithelial cells, IAV infection resulted in the activation of JNK, p38 and ERK. Inhibition of JNK and/or p38, but not ERK, led to reduced expression of RANTES/CCL5, indicating that JNK and/or p38 activation is required for chemokine expression in response to IAV infection in airway epithelial cells [51]. This is further supported by a study analysing the transcriptomic profile of human bronchial epithelial cells in response to IAV infection. The study demonstrated that among 165 upregulated genes, 29 genes (about 17.5%) were regulated by JNK and/or p38 [52]. These genes are involved in various cellular activities including antiviral activity such as MX1, antigen presentation including HLA-A and HLA-C, cell adhesion such as ICAM-1, inflammation such as IL-6 and apoptosis such as CASP10 [52], demonstrating the essential and diverse functions of these kinases in airway epithelial cells in response to IAV infections. In monocytes/macrophages, MAPKs are activated temporally in response to IAV infections. In murine monocytic cells, the influenza virus X-31 induced the activation of JNK and ERK as early as 15 min post-infection, followed by p38 activation at 3 h post-infection [53]. The early activations of JNK and ERK were important for early cytokine/chemokine responses towards the infection, which was demonstrated by the wide spread inhibition of inflammatory mediators including TNFα, IL-6, MCP-1, MIP-1α/CCL3, RANTES/CCL5, KC/CXCL1, IP10/CXCL10, and G-CSF upon inhibition of JNK and/or ERK alone [53]. In human macrophages, inhibition of JNK and ERK by their specific inhibitors or by decoy receptor 3 (DcR3) inhibited the secretion of cytokines including TNFα, IL-6 and IFNα upon IAV H1N1 infection [54]. Furthermore, it was found that sesamin, a natural compound isolated from the Thai medicinal plant Sesamum indicum, inhibited both pro-inflammatory cytokines IL-1β and TNFα in human peripheral blood mononuclear cells, by inhibiting the activation of JNK, p38 and ERK in response to H1N1 influenza infections, in a study using a combinatorial screening and computational approach [55].
The importance of MAPKs in host innate inflammatory responses against IAV infections was confirmed by in vivo studies using animal models. It was found that DcR3-transgenic mice or mice administrated with DcR3 recombinant protein had reduced disease severity and lethality upon influenza H1N1 virus infection, with reduced expression of TNFα, IL-6 and IFNα in the lungs, which was associated with reduced activation of JNK and ERK [54]. In addition, it has been shown that infections by the H9N2 influenza virus strain, which originated from swine hosts, in BALB/c mice resulted in inflammation and injury in the lung with production of pro-inflammatory cytokines including TNFα, IL-1β and IL-6. Consequently, inhibition of p38 by its specific inhibitor SB203580 caused decreased levels of TNFα, IL-1β and IL-6 and alleviation of lung injury [56]. Together, these studies demonstrate the importance of MAPKs in regulation of inflammatory mediator expressions and pulmonary inflammation in response to IAVs.
3.3. MAPKs and Cytokine Storm Induced by Highly Pathogenic Influenza Infections
Highly pathogenic IAVs (HPIAVs), such as H5N1, are capable of manipulating crucial host cell signalling, including MAPKs, to induce an overabundant expression of the inflammatory mediators. Such excessive and uncontrolled release of pro-inflammatory cytokines, such as IL-1β, IL-6 and TNFα, results in the generation of the cytokine storm that often leads to acute lung injuries and ARDS, which is a common characteristic of severe respiratory viral diseases caused by HPIAVs or other viruses including SARS-CoVs [57]. The contribution of MAPKs to the development of the cytokine storm has been documented in numerous studies (Figure 1). In response to HPIAVs infection, such as infection by H5N1, which induces deregulated pro-inflammatory cytokine expression, inhibition of c-Jun, a target molecule of JNK and a component of AP-1, has been shown to suppress the expression of IFNβ and other cytokines, including IL-6 and TNFα. Thus, suggesting that uncontrolled activation of JNK induced by highly pathogenic IAV strains may contribute to the development of cytokine storm and severe lung pathologies [58]. Activation of p38 kinase has also been shown to play an important role in cytokine hyper-induction mediated by H5N1 virus in primary human macrophages [59]. p38 inhibition using the inhibitor SB203580 resulted in >80% reduction of IFNβ transcription and significant reduction of other cytokine expression, including IFN-λ1, TNFα, MCP-1 and IP-10/CXCL10, upon H5N1 infection without affecting IRF3 nuclear translocation, indicating that the regulation of these cytokine expressions by p38 is independent of IRF3 activation [59]. The reduction rate of IFNβ transcription by p38 inhibition was comparable to that caused by IRF3 knockdown, and the combination of both resulted in an additional 14% reduction of IFNβ transcription and further reduction of cytokine expressions [59]. Studies on cytokine expression in endothelial cells in response to highly pathogenic influenza virus infection further demonstrated the crucial regulating p38 in IAV-induced cytokine deregulation [60]. It was found that more than 90% of immune/inflammatory genes induced by HPIAVs, including H7N7 and H5N1, were dependent on p38. This kinase was not only able to directly regulate the transcription of IFNβ through its promoter but, was also able to control the expression of ISGs including MxA, OAS1 and IP10 through the phosphorylation of Tyr701 and Ser727 in STAT1 [60]. Furthermore, inhibition of p38 activation nearly abolished the hyper-induction of cytokines and protected mice from lethal infection of H5N1 [60]. Taken together, these studies demonstrated a central role of p38 kinase activation in the development of the cytokine storm after induction by HPIAVs, which may be targeted for the development of therapeutic interventions for severe influenza.
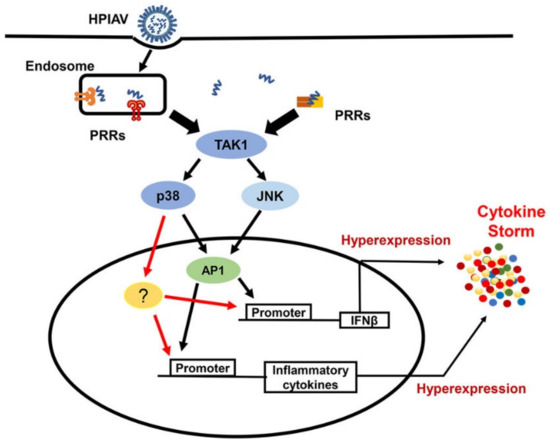
Figure 1. Pivotal Role of Mitogen-Activated Protein Kinases (MAPKs) in cytokine storm induced by highly pathogenic influenza virus infection. In response to infection by highly pathogenic influenza viruses (HPIAVs) such as H5N1 infection, PRRs, including RIG-I and TLR7 signalling, activates MAPKs, including JNK and p38, through TAK1. JNK activation contributes to hyper-expression of IFNβ and other inflammatory cytokines via transcription factor AP-1. p38, on the other hand, regulates more than 90% of inflammatory cytokine genes through AP-1 and other unknown factors. PRR—pattern recognition receptor; TAK1—TGF-beta activated kinase 1; TBK1—TANK-binding kinase 1; AP1—activator protein 1; ?—Other factors that have not been confirmed.
References
- Szymanski, C.M.; Schnaar, R.L.; Aebi, M. Bacterial and Viral Infections. In Essentials of Glycobiology, 3ed eds.; Varki, A., Cummings, R.D.; et al. Cold Spring Harbor: Long Island, NY, USA, 2015; pp. 527–538.
- Burrell, C.J.; Howard, C.R.; Murphy, F.A. Epidemiology of Viral Infections. In Fenner and White’s Medical Virology; Academic Press: Cambridge, MA, USA, 2017; pp. 185–203.
- Samji, T. Influenza A: Understanding the viral life cycle. Yale J. Biol. Med. 2009, 82, 153–159.
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses. 2011, 3, 920–940.
- Aoshi, T.; Koyama, S.; Kobiyama, K.; Akira, S.; Ishii, K.J. Innate and adaptive immune responses to viral infection and vaccination. Curr. Opin. Virol. 2011, 1, 226–232.
- Villarreal, L.P. Evolution of Viruses. In Encyclopedia of Virology; ASM press: Washington, DC, USA, 2008; pp. 174–184.
- Wang, X.; Li, Y.; O’Brien, K.L.; et al. Global burden of respiratory infections associated with seasonal influenza in children under 5 years in 2018: A systematic review and modelling study. Lancet Glob. Health 2020, 8, e497–e510.
- Kalil, A.C.; Thomas, P.G. Influenza virus-related critical illness: Pathophysiology and epidemiology. Crit. Care 2019, 23, 258.
- Peteranderl, C.; Herold, S.; Schmoldt, C. Human Influenza Virus Infections. Semin. Respir. Crit. Care Med. 2016, 37, 487–500.
- White, M.R.; Doss, M.; Boland, P.; Tecle, T.; Hartshorn, K.L. Innate immunity to influenza virus: Implications for future therapy. Expert Rev. Clin. Immunol. 2008, 4, 497–514.
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328.
- Dou, D.; Revol, R.; Östbye, H.; Wang, H.; Daniels, R. Influenza A Virus Cell Entry, Replication, Virion Assembly and Movement. Front. Immunol. 2018, 9, 1581.
- Ichinohe, T. Respective roles of TLR, RIG-I and NLRP3 in influenza virus infection and immunity: Impact on vaccine design. Expert Rev. Vaccines 2010, 9, 1315–1324.
- Wu, W.; Zhang, W.; Duggan, E.S.; Booth, J.L.; Zou, M.H.; Metcalf, J.P. RIG-I and TLR3 are both required for maximum interferon induction by influenza virus in human lung alveolar epithelial cells. Virology 2015, 482, 181–188.
- Chen, X.; Liu, S.; Goraya, M.U.; Maarouf, M.; Huang, S.; Chen, J.-L. Host Immune Response to Influenza A Virus Infection. Front. Immunol. 2018, 9, 320.
- Liu, G.; Lu, Y.; Thulasi Raman, S.N.; Xu, F.; Wu, Q.; Li, Z.; Brownlie, R.; Liu, Q.; Zhou, Y. Nuclear-resident RIG-I senses viral replication inducing antiviral immunity. Nat. Commun. 2018, 9, 3199.
- Moore, C.B.; Ting, J.P.Y. Regulation of Mitochondrial Antiviral Signaling Pathways. Immunity 2008, 28, 735–739.
- Öhman, T.; Rintahaka, J.; Kalkkinen, N.; Matikainen, S.; Nyman, T.A. Actin and RIG-I/MAVS Signaling Components Translocate to Mitochondria upon Influenza A Virus Infection of Human Primary Macrophages. J. Immunol. 2009, 182, 5682.
- Pang, I.K.; Pillai, P.S.; Iwasaki, A. Efficient influenza A virus replication in the respiratory tract requires signals from TLR7 and RIG-I. Proc. Natl. Acad. Sci. USA 2013, 110, 13910.
- Stegemann-Koniszewski, S.; Behrens, S.; Boehme, J.D.; Hochnadel, I.; Riese, P.; Guzmán, C.A.; Bruder, D. Respiratory Influenza A Virus Infection Triggers Local and Systemic Natural Killer Cell Activation via Toll-Like Receptor 7. Front. Immunol. 2018, 9, 245.
- Wu, W.; Wang, X.; Zhang, W.; Tian, L.; Leland Booth, J.; Duggan, E.S.; More, S.; Liu, L.; Dozmorov, M.; Metcalf, J.P. RIG-I Signaling via MAVS Is Dispensable for Survival in Lethal Influenza Infection In Vivo. Med. Inflamm. 2018, 2018, 6808934.
- Jeisy-Scott, V.; Kim, J.H.; Davis, W.G.; Cao, W.; Katz, J.M.; Sambhara, S. TLR7 Recognition Is Dispensable for Influenza Virus A Infection but Important for the Induction of Hemagglutinin-Specific Antibodies in Response to the 2009 Pandemic Split Vaccine in Mice. J. Virol. 2012, 86, 10988.
- Tate, M.D.; Mansell, A. An update on the NLRP3 inflammasome and influenza: The road to redemption or perdition? Curr. Opin. Immunol. 2018, 54, 80–85.
- Tate, M.D.; Ong, J.D.H.; Dowling, J.K.; McAuley, J.L.; Robertson, A.B.; Latz, E.; Drummond, G.R.; Cooper, M.A.; Hertzog, P.J.; Mansell, A. Reassessing the role of the NLRP3 inflammasome during pathogenic influenza A virus infection via temporal inhibition. Sci. Rep. 2016, 6, 27912.
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P.-Y. The NLRP3 Inflammasome Mediates In Vivo Innate Immunity to Influenza A Virus through Recognition of Viral RNA. Immunity 2009, 30, 556–565.
- Ichinohe, T.; Pang, I.K.; Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010, 11, 404–410.
- McAuley, J.L.; Tate, M.D.; MacKenzie-Kludas, C.J.; Pinar, A.; Zeng, W.; Stutz, A.; Latz, E.; Brown, L.E.; Mansell, A. Activation of the NLRP3 Inflammasome by IAV Virulence Protein PB1-F2 Contributes to Severe Pathophysiology and Disease. PLoS Pathog. 2013, 9, e1003392.
- Mubarak, R.A.; Roberts, N.; Mason, R.J.; Alper, S.; Chu, H.W. Comparison of pro- and anti-inflammatory responses in paired human primary airway epithelial cells and alveolar macrophages. Respir. Res. 2018, 19, 126.
- Allard, B.; Panariti, A.; Martin, J.G. Alveolar Macrophages in the Resolution of Inflammation, Tissue Repair, and Tolerance to Infection. Front. Immunol. 2018, 9, 1777.
- Yamada, M.; Fujino, N.; Ichinose, M. Inflammatory responses in the initiation of lung repair and regeneration: Their role in stimulating lung resident stem cells. Inflamm. Regen. 2016, 36, 15.
- Hiemstra, P.S.; McCray, P.B.; Bals, R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur. Respir. J. 2015, 45, 1150.
- Newton, A.H.; Cardani, A.; Braciale, T.J. The host immune response in respiratory virus infection: Balancing virus clearance and immunopathology. Semin. Immunopathol. 2016, 38, 471–482.
- Vareille, M.; Kieninger, E.; Edwards, M.R.; Regamey, N. The airway epithelium: Soldier in the fight against respiratory viruses. Clin. Microbiol. Rev. 2011, 24, 210–229.
- Pyle, C.J.; Uwadiae, F.I.; Swieboda, D.P.; Harker, J.A. Early IL-6 signalling promotes IL-27 dependent maturation of regulatory T cells in the lungs and resolution of viral immunopathology. PLoS Pathog. 2017, 13, e1006640.
- Seo, S.H.; Webster, R.G. Tumor necrosis factor alpha exerts powerful anti-influenza virus effects in lung epithelial cells. J. Virol. 2002, 76, 1071–1076.
- Huang, F.F.; Barnes, P.F.; Feng, Y.; Donis, R.; Chroneos, Z.C.; Idell, S.; Shams, H. GM-CSF in the lung protects against lethal influenza infection. Am. J. Respir. Crit. Care Med. 2011, 184, 259–268.
- Subramaniam, R.; Hillberry, Z.; Chen, H.; Feng, Y.; Fletcher, K.; Neuenschwander, P.; Shams, H. Delivery of GM-CSF to Protect against Influenza Pneumonia. PLoS ONE 2015, 10, e0124593.
- Halstead, E.S.; Umstead, T.M.; Davies, M.L.; Kawasawa, Y.I.; Silveyra, P.; Howyrlak, J.; Chroneos, Z.C. GM-CSF overexpression after influenza a virus infection prevents mortality and moderates M1-like airway monocyte/macrophage polarization. Respir. Res. 2018, 19, 3.
- Friesenhagen, J.; Boergeling, Y.; Hrincius, E.; Ludwig, S.; Roth, J.; Viemann, D. Highly pathogenic avian influenza viruses inhibit effective immune responses of human blood-derived macrophages. J. Leukoc. Biol. 2012, 92, 11–20.
- Zhang, J.; Liu, J.; Yuan, Y.; Huang, F.; Ma, R.; Luo, B.; Zhang, X. Two waves of pro-inflammatory factors are released during the influenza A virus (IAV)-driven pulmonary immunopathogenesis. PLOS Pathog. 2020, 16, e1008334.
- Ichikawa, A.; Kuba, K.; Morita, M.; Chida, S.; Tezuka, H.; Hara, H.; Kawaoka, Y. CXCL10-CXCR3 enhances the development of neutrophil-mediated fulminant lung injury of viral and nonviral origin. Am. J. Respir. Crit. Care Med. 2013, 187, 65–77.
- Carlin, L.E.; Hemann, E.A.; Zacharias, Z.R.; Heusel, J.W.; Legge, K.L. Natural Killer Cell Recruitment to the Lung During Influenza A Virus Infection Is Dependent on CXCR3, CCR5, and Virus Exposure Dose. Front. Immunol. 2018, 9, 781.
- Camp, J.V.; Jonsson, C.B. A Role for Neutrophils in Viral Respiratory Disease. Front. Immunol. 2017, 8, 550.
- Tate, M.D.; Brooks, A.G.; Reading, P.C. The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir. Res. 2008, 9, 57.
- Wang, J.; Nikrad, M.P.; Travanty, E.A.; Zhou, B.; Phang, T.; Gao, B.; Wentworth, D. Innate Immune Response of Human Alveolar Macrophages during Influenza A Infection. PLoS ONE 2012, 7, e29879.
- Goritzka, M.; Makris, S.; Kausar, F.; Durant, L.R.; Pereira, C.; Kumagai, Y.; Culley, F.J.; Mack, M.; Akira, S.; Johansson, C. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J. Exp. Med. 2015, 212, 699–714.
- Killip, M.J.; Fodor, E.; Randall, R.E. Influenza virus activation of the interferon system. Virus Res. 2015, 209, 11–22.
- Ludwig, S.; Ehrhardt, C.; Neumeier, E.R.; Kracht, M.; Rapp, U.R.; Pleschka, S. Influenza virus-induced AP-1-dependent gene expression requires activation of the JNK signaling pathway. J. Biol. Chem. 2001, 276, 10990–10998.
- Zhong, M.; Wang, H.; Ma, L.; Yan, H.; Wu, S.; Gu, Z.; Li, Y. DMO-CAP inhibits influenza virus replication by activating heme oxygenase-1-mediated IFN response. Virol. J. 2019, 16, 21.
- Bahadoran, A.; Lee, S.H.; Wang, S.M.; Manikam, R.; Rajarajeswaran, J.; Raju, C.S.; Sekaran, S.D. Immune Responses to Influenza Virus and Its Correlation to Age and Inherited Factors. Front. Microbiol. 2016, 7, 1841.
- Kujime, K.; Hashimoto, S.; Gon, Y.; Shimizu, K.; Horie, T. p38 Mitogen-Activated Protein Kinase and c-Jun-NH2-Terminal Kinase Regulate RANTES Production by Influenza Virus-Infected Human Bronchial Epithelial Cells. J. Immunol. 2000, 164, 3222.
- Hayashi, S.; Jibiki, I.; Asai, Y.; Gon, Y.; Kobayashi, T.; Ichiwata, T.; Shimizu, K.; Hashimoto, S. Analysis of gene expression in human bronchial epithelial cells upon influenza virus infection and regulation by p38 mitogen-activated protein kinase and c-Jun-N-terminal kinase. Respirology 2008, 13, 203–214.
- Cannon, G.; Callahan, M.A.; Gronemus, J.Q.; Lowy, R.J. Early Activation of MAP Kinases by Influenza A Virus X-31 in Murine Macrophage Cell Lines. PLoS ONE 2014, 9, e105385.
- Huang, M.-T.; Chen, S.-T.; Wu, H.-Y.; Chen, Y.-J.; Chou, T.-Y.; Hsieh, S.-L. DcR3 suppresses influenza virus-induced macrophage activation and attenuates pulmonary inflammation and lethality. J. Mol. Med. 2015, 93, 1131–1143.
- Fanhchaksai, K.; Kodchakorn, K.; Pothacharoen, P.; Kongtawelert, P. Effect of sesamin against cytokine production from influenza type A H1N1-induced peripheral blood mononuclear cells: Computational and experimental studies. In Vitro Cell Dev. Biol. Anim. 2016, 52, 107–119.
- Wei, D.; Huang, Z.H.; Zhang, R.H.; Wang, C.L.; Xu, M.J.; Liu, B.J.; Wang, G.H.; Xu, T. Roles of p38 MAPK in the regulation of the inflammatory response to swine influenza virus-induced acute lung injury in mice. Acta Virol. 2014, 58, 374–379.
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32.
- Xie, J.; Zhang, S.; Hu, Y.; Li, D.; Cui, J.; Xue, J.; Wang, M. Regulatory roles of c-jun in H5N1 influenza virus replication and host inflammation. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2014, 1842, 2479–2488.
- Hui, K.P.Y.; Lee, S.M.Y.; Cheung, C.-Y.; Ng, I.H.Y.; Poon, L.L.M.; Guan, Y.; Ip, N.Y.Y.; Lau, A.S.Y.; Malik Peiris, J.S. Induction of Proinflammatory Cytokines in Primary Human Macrophages by Influenza A Virus (H5N1) Is Selectively Regulated by IFN Regulatory Factor 3 and p38 MAPK. J. Immunol. 2009, 182, 1088.
- Börgeling, Y.; Schmolke, M.; Viemann, D.; Nordhoff, C.; Roth, J.; Ludwig, S. Inhibition of p38 mitogen-activated protein kinase impairs influenza virus-induced primary and secondary host gene responses and protects mice from lethal H5N1 infection. J. Biol. Chem. 2014, 289, 13–27.

