The therapeutic application of BMVs has largely been explored pre-clinically for its use as an immune-modulating agent
[79][80][81]. This is primarily because of the presence of antigenic protein molecules in BMVs which when used, may trigger a favorable immune response in the body. Until now this phenomenon has been successfully translated to clinics for the development of a vaccine against
Neisseria meningitidis serogroup B (Bexsero
® developed by Novartis)
[82]. In some cases, BMVs have been demonstrated to amplify the immunogenicity of a low immunogenic protein antigen by acting as a vaccine delivery system
[83][84]. Additionally, the ease of genetic modification of the bacterial source has also contributed to it being utilized as an efficient and promising immunomodulator.
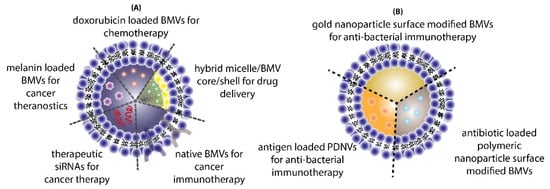
The use of BMVs for drug/cargo delivery applications has only been explored recently with only a handful of published literature reports. Most of the applications for BMVs has primarily been focused either on cancer therapy or antibacterial therapy (Figure 4).
3.1. Cancer Therapy
In one such report, BMVs isolated from non-pathogenic attenuated
K. pneumoniae were utilized for the delivery of doxorubicin
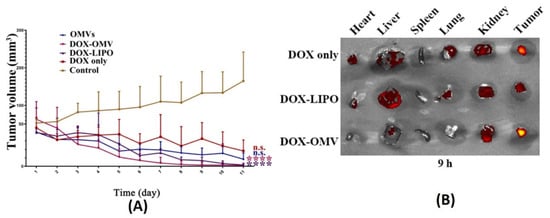
[85]. Anti-tumor studies carried out using such BMVs at a dose of 2 mg/Kg of DOX (injected intraperitoneally every day for 11 days) in A549 tumor bearing BALB/c nude mice showed a significant reduction in tumor volume as compared to the use of free drug, empty BMVs, and even doxorubicin-loaded liposomes (
Figure 5A). In fact, it was observed that the rate of reduction in tumor volumes was found to be greater for DOX-loaded BMVs as compared to DOX-loaded liposomes, signifying that BMVs showed a better therapeutic response. This added therapeutic response observed in DOX-loaded BMVs could be attributed to the favorable immune response that BMVs can induce in vivo which in conjunction with chemotherapeutic drugs leads to generation of a higher therapeutic efficacy. This was supported by tumor volume reduction studies in the same report that showed that the use of bare BMVs alone in vivo lead to a significant reduction in tumor volume as compared to untreated controls. Further, it was also observed that there occurred a significant accumulation of murine macrophages in tumor tissues that were treated with both doxorubicin loaded BMVs and empty BMVs. Pharmacokinetic analysis showed that the use of drug loaded BMVs lead to a greater drug retention in tumors that lasted for longer periods of time as compared to DOX-loaded liposomes with a concurrently lower retention found in the heart (
Figure 5B). As a result, the cardiac toxicity (which is notable in the use of doxorubicin) was found to be significantly reduced when DOX-loaded BMVs were used (as measured through analysis of lactate dehydrogenase, aspartate aminotransferase, and creatine kinase isoenzyme in blood), which were further supported through histopathological analysis of cardiac tissues. Overall, the pharmacokinetic profile of the loaded drug was improved (characterized by an increase in the drug half-life, reduction in clearance rate, and improved bioavailability) when BMVs were utilized as a drug delivery vehicle. Immunotoxicity analysis in C57BL/6 normal mice at the therapeutic dosage (over a period of 11 days) showed that the administration of both DOX-loaded BMVs and bare BMVs lead to a significant increase in serum cytokine levels which returned back to basal levels over a period of time. These results therefore showed that BMVs could be well tolerated in vivo and could be used as an effective drug delivery vehicle.
Figure 5. (
A) Tumor volume reduction measurements and (
B) in vivo drug distribution of doxorubicin-loaded
K. pneumoniae OMVs as compared to controls. Reproduced from
[85], Elsevier, 2020.
Similar to the above, a study depicting the use of
S. typhimurium BMVs for combined drug delivery and immunotherapy against cancer was reported
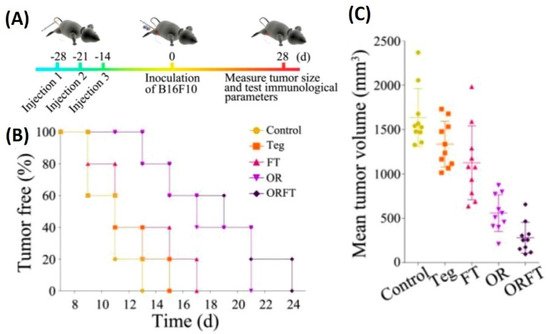
[86]. In vivo tumor therapy studies carried out using hybrid BMVs (BMV/micelle/drug) in B16F10 melanoma and 4T1 mammary tumor in C57BL/6 mice at a dose of 30 µg of BMVs (once/3 days for a total of 3 injections) lead to a significant reduction in tumor volume and increase in survival as compared to controls. Furthermore, this treatment also limited the spread of cancer metastatic nodules in lung tissues, which otherwise are prevalent in the B16F10 tumor model, which could explain the increase survival of mice observed on treatment. Interestingly, the synthesized BMVs also showed an immunoprotective effect against tumor. Mice that were pretreated with BMVs, when challenged with tumor cells later on, showed a delayed tumor growth response with significantly small tumor volumes (
Figure 6). Even though the exact reasons for the same have not been elucidated in this report, these results show overall the promising effect that BMVs have towards developing a strategy for tumor prevention and treatment. Upon BMV administration, the in vivo cytokine analysis of blood and tumor samples showed that even though there occurred an increase in the cytokine levels of TNF-α, IFN-γ, IL-12, IL-4, and IL-17, the levels of these cytokine reduced to basal levels after 24 h. Overall, no blood toxicity and organ toxicity (including liver and renal functions) were found upon BMV administration.
Figure 6. (
A) Treatment timeline for evaluating the protective role of hybrid BMVs (drug-loaded micelles surface modified with BMVs)—hybrid BMVs were administered to mice before tumor challenge. (
B) Percentage of tumor-free mice after tumor challenge (
C) Mean tumor volume measurements of hybrid OMVs against controls (Teg-Tegafur, FT-Tegafur loaded F127 micelle, OR-RGD functionalized BMV, ORFT—Tegafur-loaded F127 micelle surface modified with RGD functionalized BMV). Reproduced with permission from
[86], American Chemical Society, 2020.
Even though the above studies showed the ability of BMVs as a promising drug delivery agent, an interesting study has demonstrated its ability to be used as in its native form as a potential anti-tumor immunotherapeutic agent
[87]. To demonstrate its applicability, unmodified BMVs isolated from both Gram-negative and -positive bacterial species were assessed for their ability to actuate an anti-tumor immune response in different tumor models in mice. Specifically, BMVs isolated from Gram-negative
E. coli and
S. Enterica and Gram-positive
S. aureus and
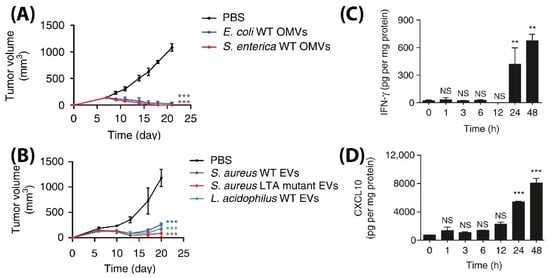
L. acidophilus were injected intravenously in BALB/c mice bearing CT26 colon adenocarcinoma at a 5 µg BMV dose (4 times at 3 days interval), significant tumor volume reductions were observed as compared to PBS injected controls (
Figure 7A,B). Additionally, to demonstrate its diverse potential, BMVs from
E. coli were assessed for their therapeutic response in CT26 colon adenocarcinoma and 4T1 mouse mammary tumor of BALB/c mice, and MC38 mouse colon adenocarcinoma and B16BL6 mouse melanoma cancer of C57BL/6 mice. At a 5 µg
E. coli BMV dose injected intravenously (4 times at 3 days interval), significant tumor volume reductions were observed for all the treated tumor types. However, the reduction in tumor volumes in 4T1 and B16BL6 tumors were found to be less effective as compared to those observed in CT26 and MC38 tumors, which shows the important role tumor biology and characteristics have on the net therapeutic outcome. Interestingly, for the treatment of CT26 tumors, a long-term memory effect was observed for treatment using
E. coli BMVs wherein secondary and tertiary challenges of CT-26 tumor cells were rejected in mice that recovered from the primary tumor challenge post BMV administration. These results show how BMVs have the ability to favorably modulate the immune system and possibly provide a protective environment to prevent tumor relapse as observed for other immunotherapeutic modalities
[88]. Similar to other reports on the use of BMVs in vivo, an increase in the levels of cytokines and chemokines such as IL-12p40, IFN-γ, CXCL10, TNF-α, IL-6, and IL-12p70 were also observed in this study. However, it was observed that the cytokines CXCL10 and IFN-γ specifically showed elevated levels in the tumor tissues over 24 and 48 h which could imply that these cytokines play an important role in eliciting an anti-tumor immune response (
Figure 7C,D).
Figure 7. Tumor volume measurement study for CT26 tumors injected with (
A) Gram-positive
S. Aureus, lipoteichoic acid
S. Aureus mutant, and
L. Acidophilus. (
B) Gram-negative
E. coli and
S. enterica. Measurement of (
C) IFN-γ and (
D) CXCL10 cytokine levels in tumor cell lysate at different time points after
E. coli BMV administration in CT26 tumors (**
p < 0.01, and ***
p < 0.001). Reproduced with permission from
[87], Springer Nature, 2017.
Apart from utilizing BMVs for drug loading, BMVs have also been utilized for the delivery of therapeutic nucleic acids for the treatment of cancer. By loading siRNA targeting kinetic spindle protein, a protein essential for spindle formation and continuation of cell cycle, into BMVs obtained from Δ
msbB E. coli, in vivo treatment of liver cancer was demonstrated
[89]. Significant reduction of HCC-1954 xenografts in nude mice was observed upon intravenous administration of siRNA-loaded BMVs at a 4 µg dose siRNA injected on alternate days over a 22-day treatment period as compared to controls. Serum cytokine analysis showed elevated levels of TNF alpha, IL6, and IFNγ in C57BL/6 mice upon repeated dosing (at 10–20 µg siRNA) over 4 consecutive days. However, this elevation was observed only for a brief period of 3 h post-administration and would return back to basal levels in 24 h. Note that lethal dose toxicity studies showed that the BMVs obtained from Δ
msbB E. coli did not cause any mortality even at a single high dose of 100 µg, while, on the other hand, administration of 50 µg of BMVs obtained from wild type
E. coli lead to mortality within 48 h post administration. This shows how the biochemical composition of the surface moieties of BMVs play a crucial role in its toxicity response. Here, the Δ
msbB mutation in
E. coli produces underacylated LPS which shows reduced endotoxicity.
While BMVs show great promise as a drug delivery vehicle, one report has gone further to evaluate its potential as a stimuli-responsive multifunctional theranostic agent
[90]. To do so, BMVs synthesized from Δ
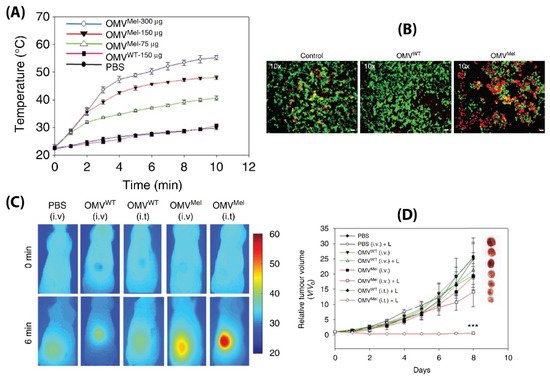
msbB E. coli were loaded with melanin (which can act both as a photoacoustic and photothermal agent), by introducing the required plasmid to the bacteria. This led to the generation of melanin which is packed within the BMV lumen. Upon laser exposure, a concentration dependent thermal response was observed for melanin loaded BMVs, which when used in vitro produced significant cell death due to the photothermal effect (
Figure 8A,B). When such melanin-loaded BMVs were administered in 4T1 mammary tumor-bearing FOX-N-1 nude mice intravenously at a single dose of ~150 µg protein, optoacoustic signals could be observed in tumor, liver, and kidneys, enabling the study of its biodistribution profile. The imaging results demonstrated that these BMVs accumulated in the tumor through EPR effects, underwent continuous circulation in vivo, and cleared slowly from the system over a period of 24 h. Photothermal treatment of the 4T1 tumors 3 h post-injection of a single dose of ~75 µg BMVs lead to a significant thermal response of 56 °C and 47 °C for intratumoral and intravenous administration respectively. This resulted in a significant reduction in tumor volume over an 8 days period after just a single treatment and laser therapy which its high effectiveness in cancer therapy (
Figure 8C,D). As previous studies have pointed out, there occurred a significant increase in the cytokine levels of TNF-α, IL-6, and IFN-γ 2 h post administration of BMVs, which however reduced near to the baseline levels after 25 h.
Figure 8. (
A) Thermal response of melanin-loaded OMVs at different concentrations as compared to wild type OMVs upon laser exposure. (
B) Live–dead (calcein AM/EthD-1) staining of 4T1 cancer cells treated with melanin-loaded OMVs and exposed to laser as compared to controls. Thermal imaging (
C) and tumor volume reduction measurements (
D) of 4T1 tumors after either intravenous or intratumoral administration of melanin loaded OMVs and laser exposure as compared to controls. Reproduced with permission from
[90], Springer Nature, 2019.
3.2. Antibacterial Therapy
The use of BMVs for antibacterial applications has mostly been investigated by utilizing the inherent antigenic molecules present on BMV surface for eliciting a favorable immune response against the invading pathogen. In this regard, BMVs have been utilized either directly in its unmodified form
[91] or modified appropriately to present the antigenic proteins with a suitable material to elicit a desired immune response. One way of presenting antigenic proteins to the immune system is through the use of NPs, which can maximize immune cell recognition owing to their large surface area and size scale that facilitates particle uptake
[92][93][94]. Numerous literature reports have utilized this methodology to develop nanotechnology-based vaccines and similar approaches have been demonstrated with BMVs for antibacterial therapy.
To demonstrate the potential of NP-based immunomodulation by utilizing BMV antigen proteins, surface-modified Au NPs have been investigated in vivo for antibacterial therapy applications
[95]. Upon intravenous administration of BMV protein-coated Au NPs (2.5 µg dose) into immunocompetent CD-1 mice, a number of highly precise immune responses were observed as compared to bare BMVs and PBS injected controls. Specifically, it was observed that BMV-coated Au NPs lead to a heighted activation and maturation of dendritic cells and T cells and an increased B cell response and a consequent increase in antibody titers. Note, in this study, that when smaller sized Au NPs (~30 nm) were used as compared to larger sized Au NPs (~90 nm) a relatively heighted dendritic cell activation and maturation was observed. This was found to be due to the better accumulation of smaller sized Au NPs in the lymph nodes of mice, thereby making them more suitable for immune activation. However, in this study the application of these BMV protein surface-modified Au NPs was not evaluated in an infection model.
In an interesting report, BMVs were utilized to surface modify polymeric NP-based antibiotic delivery systems such that target specificity could be achieved
[96]. Preliminary in vitro studies undertaken by this group showed that BMVs isolated either from
E. coli or
S. aureus showed specific uptake in macrophages that were pre-infected with
E. coli or
S. aureus, respectively. As a result, antibiotic-loaded PLGA NPs surface modified with
S. aureus BMVs were found to show significantly greater accumulation in major organs of an
S. aureus infected BALB/c mice model as compared to
E. coli BMV-coated PLGA NPs and liposome-coated PLGA NPs. This greater internalization was explained to be caused by the greater ability of infected macrophages to internalize the NPs first, followed by the natural biodistribution of these macrophages to the infected organs. As a result of this greater accumulation observed for the
S. aureus BMV-coated antibiotic loaded PLGA NPs, effective reduction of bacterial CFU counts could be observed in kidneys and lungs of the infected mice. However, note here that effective therapeutic efficacy could not be achieved for all major organs, and more studies need to be carried out to exploit this specific property of BMV-coated NPs. It would be interesting to study in this case if the specific uptake of BMV-coated NPs in infected macrophages could be extended to other infected mammalian cells and cancer cells. If such a specificity could be achieved, it could open up the possibility of treating patients with tumors bearing bacterial load. Such infected tumor conditions are nowadays observed in clinical investigations and the presence of these bacteria in tumors are found to play an important role in inhibiting the efficacy of chemotherapeutic drug treatments
[97][98].
Beyond the use of BMVs, one unique report demonstrated the direct use of bacterial cells for NP surface modification
[99]. Here, bacterial protoplasts were first isolated by treating bacteria with lysozyme to remove the bacterial cell wall, followed by its serial extrusion through 10, 5, and 1 µm sized polycarbonate membrane filters. These protoplast-derived nanovesicles (PDNVs) show inherent advantage over BMVs as they can be directly synthesized in large amounts from the bacterial suspension. Additionally, due to removal of the bacterial cell wall, the resultant PDNVs were depleted of the outer membrane proteins OmpA and lipid A which are components of LPS, thus making them less immunogenic and more favorable for drug delivery and theranostic applications. Here, for investigating its immunotherapeutic potential, PDNVs were loaded separately either with
E. coli antigen OmpA and
S. aureus antigen Scoagulase by expressing the desired antigen in the parent
E. coli bacterium. The resultant PDNVs, when administered in vivo in C57BL/6 mice, showed high specificity in developing an immune-protective response against bacterial challenge. Specifically, it was observed that PDNVs harboring either the OmpA antigen or the Scoagulase antigen showed effective immune response in vivo when challenged with lethal doses of
E. coli or
S. aureus respectively. This response was found to be highly specific with respect to the antigen that the PDNVs harbored such that mice that were administered with PDNVs harbouring
E. coli antigen OmpA succumbed when challenged with
S. aureus infection, and vice versa. The protective immunity offered by such PDNVs could be observed in mice up to 6 weeks post-administration. Interestingly, these PDNVs were found to show low in vitro and in vivo toxicity as compared to
E. coli-derived BMVs. Specifically, it was observed that even upon administration of upto 1 mg of PDNVs in C57BL/6 mice all animals survived, while a dosage of only 25 µg of BMVs lead to the death of 80% of the animals. Overall, PDNVs show great promise towards the development of biomimetic drug delivery vehicles and show several advantages as compared to BMVs.
 +1 credit
+1 credit