 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Xiaowei Xu | + 2578 word(s) | 2578 | 2021-09-12 05:24:21 | | | |
| 2 | Conner Chen | Meta information modification | 2578 | 2021-09-24 10:38:32 | | |
Video Upload Options
Proteomics is a kind of omics which studies the protein composition, distribution and changing rules in cells, tissues or organisms. Essentially, it refers to the macroscale study of protein characteristics, including protein expression level, post-translational modification, small molecule–protein interaction and so on. Research on the proteome cannot only provide the material foundation for the law of the activities of life, but also provides a theoretical foundation and solutions to elucidate and conquer numerous types of mechanisms of illness. Click chemistry was first put forward by K B Sharpless in 2001 which provides a quick and reliable synthesis method for different molecules to offer a range of reactivities, orthogonality and utility in various applications. Click chemistry is characterized by good chemical selectivity, favorable solvent compatibility, diverse modularization, minimum synthesis requirements and high yield, upon which it considerably reduces the effect of sensor incorporation on protein activity and reveals the structure and functionality of proteins.
1. Activity-Based Protein Profiling
Over the past few years, activity-based protein profiling (ABPP) has become a strong method of chemical proteomics for analyzing proteins’ functional states within a complex proteome [1][2]. ABPP strategies usually use activity-based probes (ABPs), that are designed to be recognized by the target protein and react with residues from the active site, which can efficiently enrich and identify of low-abundance and low-affinity probe-interacting proteins [3]. Many ABPs have been developed for many classes of enzymes, including serine hydrolases [4][5], cysteine proteases [6], metallohydrolases, phosphatases, deubiquinating enzymes [7][8], kinases [9][10], various oxidoreductases, and others. Although ABPs often utilize reporter groups for the direct enrichment or visualization of labeled proteins, avoiding the need for additional conjugation steps, such bulky groups can hamper cellular uptake and tissue distribution, potentially limiting their application in living systems. To improve these problems, compound-centric chemical proteomics approach on the basis of click reactions were used to enrich and identify enzyme targets, which allow for the incorporation of chemical groups with highly selective reactivity into small molecules, or protein modifications without perturbing their biological function, enabling the selective installation of an analysis tag for downstream investigations (Figure 1). The probe is designed based on the structure of the active molecule, then added to live cells or tissues. It reacts covalently (via an electrophilic trap or a photo-crosslinking group) or non-covalently with the target protein. The lysed samples subject to CuAAC reaction attach a fluorophore, affinity label, or a combination of these elements. Marked proteins are subsequently visualized, identified or quantified using a variety of techniques, such as SDS-PAGE, LC-MS/MS analysis or confocal imaging [3]. The CuAAC-enabled ABPs were developed for protein arginine deiminases [11]; ubiquitin mechanisms [7][12], cytochrome P450 enzymes [13], glycosidases [14], and kinase [15][16].
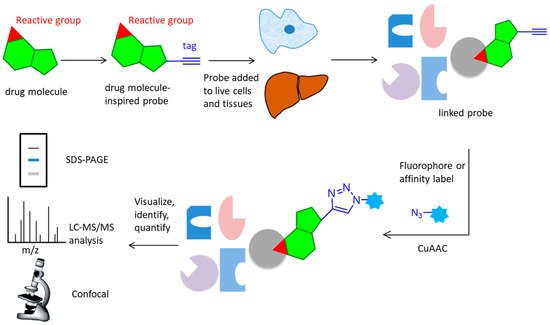
Figure 1. Overall workflow of the activity-based protein profiling approach. A probe with an active group and an alkynyl tag reacts with the active site of target protein in live cells and tissues, then undergoes CuAAC reaction with an azide containing fluorophore or affinity label. After lysis and separation, the probe is fluorescently scanned in SDS-PAGE display, LC-MS/MS analysis, or confocal imaging for identification or quantification.
Clickable probes based on light affinity can be the most common chemical proteomics tool for capturing and identifying non-covalent targets for small bioactive molecules [17]. These probes include the synthesis and installation of a click group (azide or alkine) and photoaffinity groups (such as diazirines, benzophenone) to cause as little disruption as possible to the biological activity of the compounds, which sometimes necessitates a thorough SAR investigation and even new synthetic channels [18][19][20]. Cell permeable sensors with both clickable group and light affinity have been developed to characterize a variety of clinically approved medications and inhibitors, including kinase inhibitors [21], γ-secretory enzyme inhibitors [22], β-secretor inhibitors [23], antibiotics [24], NSAIDs [25], epigenetic regulatory compounds [26], natural products [27], and also protein interactions with lipids [28] and sterol [29]. Considering that metabolites may participate in functional sites of proteins, the metabolite-derived click probes can be used as a valuable analytical tool to plot drug interactions in the proteomes, and even as a tool for uncovering functional regulators of metabolite binding proteins [28].
As a carbocyclic analog of adenosine, 3-Deazaneplanocin A (DzNep) can inhibit the activity of histone lysin methyltransferase, which has aroused great interest in epigenetic research over the past few years [30][31][32]. However, the molecular mechanism and extracellular targets of DzNep have not been fully understood. Yang et al. developed some small-molecular probes derived from DzNep that are permeable to the cells, but the bulky modification groups in the probes usually disrupt the interactions between proteins and drugs, so it is still challenging to efficiently capture cell targets in situ interactions [21][33][34][35]. Therewith, Tam and colleagues designed a novel “clickable” affinity-based probe DZ-1, with minimal structural modification from DzNep (Figure 2). DZ-1 possessed comparative anti-apoptotic activity as DzNep in MCF-7 mammalian cells. In situ proteome profiling of DZ-1 was successfully carried out on the basis of pull-down LC-MS/MS analysis. Finally, some highly enriched proteins were identified as potential cellular protein targets of DzNep [36].
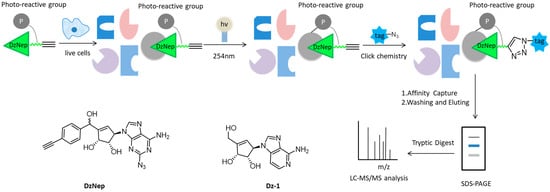
Figure 2. Overall workflow of the cell-based proteome profiling approach followed by large-scale pull-down/LC-MS/MS for identification of potential cellular targets of DzNep using affinity-based probe DZ-1.
Inflammation-related processes are pivotal factors contributing to sepsis-associated cardiac dysfunction. Cardiac neutrophil infiltration and subsequent release of myeloperoxidase leads to the formation of the oxidant hypochlorous acid (HOCl) that is able to chemically modify plasmalogens into 2-chlorohexadecanal (2-ClHDA). To elucidate this metabolic process and characterize protein targets for 2-ClHDA, a clickable alkynyl analog, 2-chlorohexadec-15-in-1-al (2-ClHDyA), was used by Prasch and colleagues to identify its protein targets (Figure 3a). Through CuAAC reaction of 5-tetramethylrhodamine azide (N3-TAMRA) and two-dimensional gel electrophoresis, they were able to pinpoint 51 proteins which form adducts with 2-ClhdYA. Genetic ontology enrichment analysis showed that heat shock and chaperone proteins, energetic metabolism and cytoskeleton proteins were the key targets for HOCl modified lipids in the heart of mice with endotoxemia [37].
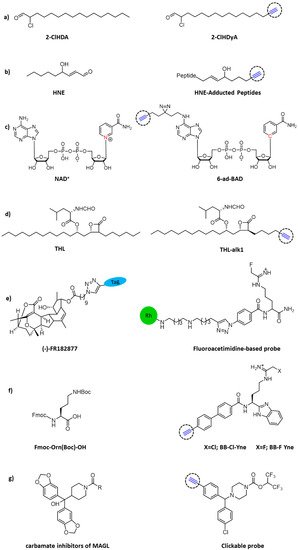
Figure 3. Chemical structures of clickable probes in ABPP for identifying protein targets of: (a) 2-ClHDA, (b) HNE, (c) NAD+, (d) THL, (e) natural product (-)–FR182877, (f) Cl- and F-amidine, and (g) carbamates.
Diffusible electrophilic α, β-unsaturated aldehydes, such as 4-hydroxynonenal (HNE), are primary targets of free radical damage during oxidative stress. Some studies have shown that HNE or other electrophilic agents can modify IκB kinase (IKK) [38], tubulin isomer [39], and Keap1 [40][41], leading to the loss of protein function and disturbance of cell signal transmission. In order to fully understand the effect of oxidative stress on cellular signaling transduction and disease pathology, it is necessary to analyze HNE modified proteins in vivo. Vila and colleagues explored Staudinger’s ligation and CuAAC to selectively label proteins with HNE in colon cancer cells, and subsequently pull-down by biotin–streptavidin interaction for LC-MS/MS analysis (Figure 3b). The results showed that both strategies produced effective biotinylation of HNE-conjugated protein, while click chemistry was proven to be superior for recovering proteins [42].
Nicotinamide adenine dinucleotide (NAD+), known as oxidoreductase coenzyme, is also a multifunctional substrate of many post-translational modification enzymes, such as poly-ADP−ribose polymerases (PARP) and sirtuins [43]. The recent studies of noncanonical NAD-binding proteins suggest that powerful chemical tools for profiling the NAD interactome are quite necessary. Šileikytė and colleagues developed a NAD+/NADH probe, 6-ad-BAD, with two reactive sites for both click reaction and light crosslinking. Moreover, the nicotinamide linked to ribose was replaced by a benzamide adenine dinucleotide (BAD) to avoid enzyme digestion (Figure 3c). Results showed that 6-ad-BAD could label PARP effectively in a UV dependent manner. Then, the chemical proteomics of 6-ad-BAD was evaluated in HEK 293T cell lysate through biotinylated enrichment and 24 unknown NAD or related nucleotide binding proteins were identified. This clickable probe will be useful in future chemical proteomics studies for profiling the NAD+ interactome across different tissues as well as in disease contexts [44].
Tetrahydrolipstatin (THL, also known as Orlistat) is an FDA approved anti-obesity drug with potential bactericidal activity. To explore the enzymatic targets of this β-lactam ring in a complex bacterial proteome, Ravindran and Wenk designed a functional THL–alkyne analog (Figure 3d) to quantify the lipid esterase activity and enriched the target proteins in Mycobacterium bovis BCG at different physiologic states [45].
Adam and colleagues used click chemistry as a handy binding method to synthesize both rhodamine-, and rhodamine and biotin-tagged (trifunctional) sensors from the natural product (-)–FR182877 [46]. Using this sensor, the researchers identified carboxyesterase-1 as the protein target in the heart of the mouse (Figure 3e). In the same way, Thompson et al. used click chemistry to covalently label rhodamine with inhibitors for arginine deiminase 4 (PAD4), an enzyme controlled by calcium [47].
Citrullination is the post-translational hydrolysis of peptidyl-arginine to form peptidyl-citrulline [48][49][50], which is a reaction also catalyzed by PADs. Abnormal increase of citrullinated protein is associated with autoimmune illnesses and cancers [48][51]. Cl-amidine and F-amidine were reported to permanently inhibit PADs by covalently altering an active cysteine site [52][53][54]. Herein, Nemmara and colleagues developed cell permeable and “clickable” probes (BB-Cl-Yne and BB-F-Yne) for covalent labeling of the PADs both in vitro and in cell-based systems [4]. These sensors covalently alter the conserved cysteine residues in all PAD isozymes and serve as the base for azide-alkyne cycloaddition, and, subsequently, CuAAC [55] with either TAMRA-N3 or biotin-N3. It is worth noting that such compounds may be used in several forms, including the off-target recognition of the parent compounds and ABPPs on the target binding tests, to demonstrate PAD inhibitor efficacy.
Chang and colleagues used a combination of competitive and click chemistry ABPP (Figure 3g) to study a variety of proteomic reactions to activate carbamates in vitro and in vivo. They identified several carbamates derivatives, among them, O-aryl and O-hexafluoroisopropyl (HFIP) carbamates could react selectively with serum hydrolases in vivo. They used the proteomic specificity of carbamate HFIP to plot in situ images of monoacylglycerol lipase endocannabinoid hydrolases and α-β hydrolase-6. They proved that carbamates are the preferred reaction group of serine hydrolase, which can adapt to different structural modifications and produce inhibitors with special potency and selectivity in the mammalian proteome [56].
Designed to identify chemical probes for functionally related protein families that can be used in complex natural environments, ABPP is an area that benefits from click chemistry. Due to the modular and efficient properties of the click reaction, the synthesis of probe libraries has been greatly simplified. However, there is still lack of a universal chemical probe or small molecule ligand for all target proteins, and the real-time dynamic imaging of a specific protein of interest in a live organism remains highly challenging. Due to the diversity of biological applications, there is no standardized protocol covering the different aspects of probe preparation, click reaction conditions, or analysis. This results in various methods that may seem overwhelming to the newcomer. Click reactions can be readily integrated to both conventional biological techniques such as gel-based fluorescent-labeling, biotin-based pull-down assay, and many of the upcoming high-throughput bioassays and characterization techniques such as microarray, LC-MS/MS, etc. Furthermore, click modules enable the enrichment of protein targets, yet their presence can sometimes perturb molecular interactions and biological activity. Recent advances in ‘‘label-free’’ proteomic methods, such as thermal proteome profiling or drug affinity responsive target stability, represent complementary strategies to plot small molecule–protein interactions, bypassing the requirement of enrichment handles.
2. Enzyme-Inhibitors Screening
Target-guided synthesis (TGS) is mainly divided into dynamic combinatorial chemistry and kinetically controlled TGS. Tethering and in situ click chemistry are representative strategies, respectively. The former method is based on thiol-disulfide exchange, in which free sulfhydryl groups on the protein surface react with small fragments containing disulfide bonds to form disulfide bonds [57], the latter uses irreversible click reactions to synthesize two reaction building blocks into a potentially inhibiting compound [58].
Traditional drug development usually depends on pharmaceutical chemistry, whether in the very beginning of drug discovery or the subsequent stages of drug optimization. Many enzymes have multiple binding domains, and apart from the active center, the allosteric binding sites mainly confer selectivity and potency [59]. In this case, click chemistry is considered to be a convenient method for assembling fragment-based inhibitors because of its highly modular and efficient reaction characteristics. As illustrated in Figure 4a, the combinations of M+N fragments result in potential bidentate inhibitor library (M×N compounds) for high-throughput screening [60].
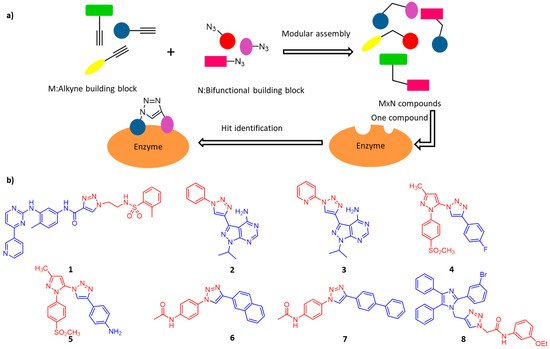
Figure 4. (a) Illustration of in situ click chemistry contributes to rapid assembly of potential bidentate inhibitors (M × N compounds) from N number of alkynes and M number of azides. (b) Potent and selective click-inhibitors of 1: Abl kinase, 2 and 3: PfPK7, 4 and 5: COX-2, 6 and 7: OGT, and 8: α-Glucosidase.
2.1. Protein Kinases
Protein kinases catalyze the phosphorylation of serine, threonine, tyrosine and histidine residues of proteins. Aberrant kinases expressions are involved in numerous illnesses, including inflammation and cancers [61]. Kalesh and colleagues recently used click chemistry to produce 344 Abelson tyrosine kinase (Abl) inhibitors [62]. Later inhibition screening assays showed that Abl kinase had a preference for short chain azide scaffolds, then 11 lead compounds with moderate potency were found. Among them, compound 1 is the most potent hit during screening with IC50 = 700 nM (Figure 4b). Similarly, Klein and colleagues used this strategy to produce Plasmodium falciparumprotein kinase 7 (PfPK7) inhibitors [63]. The researchers used alkyne/azide-derivatized purine analogs to click on various aromatic azides/alkynes, and subsequent inhibition screening assays resulted in two potent PfPK7 inhibitors (compound 2 and 3, Figure 4b) with IC50 at 10–20 μM.
2.2. Cyclooxygenase-2
Cyclooxygenase (COX) catalyzes the transformation of arachidonic acid into prostaglandins, which plays an important role in human physiology and pathological conditions [64]. Among the three subtypes of COX, COX-2 is considered to be closely related to various pathological processes, so the development of selective COX-2 inhibitors is a major focus of pharmaceutical research. Bhardwaj and colleagues demonstrated the use of the COX-2 binding site as a reaction vessel to produce its own potent and selective inhibitors. They have designed and synthesized a series of pyrazole-based azide building blocks and a series of corresponding triazole-containing biheterocyclic compounds through the in situ click chemistry method, and screened out compounds 4 and 5, (Figure 4b), which are highly effective inhibitors of COX-2 [65].
2.3. O-GlcNAC Transferase
O-GlcNAC transferase (OGT) is a critical enzyme involved in the dynamic O-GlcNAcylation of nucleoproteins and cytoplasmic proteins. The discovery of cell permeability OGT inhibitors is of great significance to elucidate the function and regulatory mechanism of O-GlcNAcylation [66]. Wang et al. combined the advantages of tethering and in situ click chemistry to find OGT inhibitors. They reported two cell permeable OGT inhibitors (compounds 6 and 7, Figure 4b), both of which significantly inhibited intracellular O-GlcNAcylation without side-effects on cell viability. Unusual non-competitive inhibition of OGT was helpful to find new inhibitors and explore the regulatory mechanism of OGT [67].
2.4. α-Glucosidases
The family of enzymes α-Glucosidases play an important role in carbohydrate digestion in vivo [68]. Inhibition of α-glucosidase activity could reduce the level of plasma glucose after surgery, therefore it has been considered as an important target for the treatment of type II diabetes mellitus [69][70]. Wang and colleagues synthesized a series of 2,4,5-triarylimidazole-1,2,3-triazole derivatives using CuAAC and evaluated their inhibitory effects on α-glucosidase. Among them, a new type of structural α-glucosidase inhibitor (compound 8, Figure 4b) was identified, which can be used as a lead compound for further development of α-glucosidase inhibitors [71].
The development of enzyme inhibitors is another important area where click chemistry plays a positive role. It is considered to be a convenient fragment-based inhibitor assembly strategy, in which a large number of potential bipedal inhibitors are generated with minimum synthetic effort. Ingenious strategies such as in situ click chemistry have so far shed some light on new ways of producing potent inhibitors against certain enzymes. However, this strategy is still in its infancy, requires a large amount of protein, and the amplification effect is relatively poor except in some highly optimized conditions.
References
- Evans, M.J.; Cravatt, B.F. Mechanism-Based Profiling of Enzyme Families. Chem. Rev. 2006, 106, 3279–3301.
- Uttamchandani, M.; Li, J.; Sun, H.; Yao, S.Q. Activity-based protein profiling: New developments and directions in functional proteomics. ChemBioChem 2008, 9, 667–675.
- Willems, L.I.; van der Linden, W.A.; Li, N.; Li, K.-Y.; Liu, N.; Hoogendoorn, S.; van der Marel, G.A.; Florea, B.I.; Overkleeft, H.S. Bioorthogonal Chemistry: Applications in Activity-Based Protein Profiling. Acc. Chem. Res. 2011, 44, 718–729.
- Nemmara, V.V.; Subramanian, V.; Muth, A.; Mondal, S.; Salinger, A.J.; Maurais, A.J.; Tilvawala, R.; Weerapana, E.; Thompson, P.R. The Development of Benzimidazole-Based Clickable Probes for the Efficient Labeling of Cellular Protein Arginine Deiminases (PADs). ACS Chem. Biol. 2018, 13, 712–722.
- An, H.; Statsyuk, A.V. Development of activity-based probes for ubiquitin and ubiquitin-like protein signaling pathways. J. Am. Chem Soc. 2013, 135, 16948–16962.
- Wright, A.T.; Cravatt, B.F. Chemical proteomic probes for profiling cytochrome p450 activities and drug interactions in vivo. Chem. Biol. 2007, 14, 1043–1051.
- Tsai, C.S.; Yen, H.Y.; Lin, M.I.; Tsai, T.I.; Wang, S.Y.; Huang, W.I.; Hsu, T.L.; Cheng, Y.S.; Fang, J.M.; Wong, C.H. Cell-permeable probe for identification and imaging of sialidases. Proc. Natl. Acad. Sci. USA 2013, 110, 2466–2471.
- Bantscheff, M.; Eberhard, D.; Abraham, Y.; Bastuck, S.; Boesche, M.; Hobson, S.; Mathieson, T.; Perrin, J.; Raida, M.; Rau, C.; et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 2007, 25, 1035–1044.
- Patricelli, M.P.; Szardenings, A.K.; Liyanage, M.; Nomanbhoy, T.K.; Wu, M.; Weissig, H.; Aban, A.; Chun, D.; Tanner, S.; Kozarich, J.W. Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry 2007, 46, 350–358.
- Lapinsky, D.J.; Johnson, D.S. Recent developments and applications of clickable photoprobes in medicinal chemistry and chemical biology. Future Med. Chem. 2015, 7, 2143–2171.
- Li, Z.; Hao, P.; Li, L.; Tan, C.Y.; Cheng, X.; Chen, G.Y.; Sze, S.K.; Shen, H.M.; Yao, S.Q. Design and synthesis of minimalist terminal alkyne-containing diazirine photo-crosslinkers and their incorporation into kinase inhibitors for cell- and tissue-based proteome profiling. Angew. Chem. Int. Ed. Engl. 2013, 52, 8551–8556.
- Li, Z.; Wang, D.; Li, L.; Pan, S.; Na, Z.; Tan, C.Y.; Yao, S.Q. “Minimalist” cyclopropene-containing photo-cross-linkers suitable for live-cell imaging and affinity-based protein labeling. J. Am. Chem. Soc. 2014, 136, 9990–9998.
- Li, J.; Wang, J.; Wen, L.; Zhu, H.; Li, S.; Huang, K.; Jiang, K.; Li, X.; Ma, C.; Qu, J.; et al. An OGA-Resistant Probe Allows Specific Visualization and Accurate Identification of O-GlcNAc-Modified Proteins in Cells. ACS Chem. Biol. 2016, 11, 3002–3006.
- Ballard, T.E.; Murrey, H.E.; Geoghegan, K.F.; am Ende, C.W.; Johnson, D.S. Investigating γ-secretase protein interactions in live cells using active site-directed clickable dual-photoaffinity probes. MedChemComm 2014, 5, 321–327.
- Zuhl, A.M.; Nolan, C.E.; Brodney, M.A.; Niessen, S.; Atchison, K.; Houle, C.; Karanian, D.A.; Ambroise, C.; Brulet, J.W.; Beck, E.M.; et al. Chemoproteomic profiling reveals that cathepsin D off-target activity drives ocular toxicity of beta-secretase inhibitors. Nat. Commun. 2016, 7, 13042.
- Eirich, J.; Orth, R.; Sieber, S.A. Unraveling the protein targets of vancomycin in living S. aureus and E. faecalis cells. J. Am. Chem. Soc. 2011, 133, 12144–12153.
- Gao, J.; Mfuh, A.; Amako, Y.; Woo, C.M. Small Molecule Interactome Mapping by Photoaffinity Labeling Reveals Binding Site Hotspots for the NSAIDs. J. Am. Chem. Soc. 2018, 140, 4259–4268.
- Montgomery, D.C.; Sorum, A.W.; Meier, J.L. Chemoproteomic profiling of lysine acetyltransferases highlights an expanded landscape of catalytic acetylation. J. Am. Chem. Soc. 2014, 136, 8669–8676.
- Wright, M.H.; Sieber, S.A. Chemical proteomics approaches for identifying the cellular targets of natural products. Nat. Prod. Rep. 2016, 33, 681–708.
- Niphakis, M.J.; Lum, K.M.; Cognetta, A.B., 3rd; Correia, B.E.; Ichu, T.A.; Olucha, J.; Brown, S.J.; Kundu, S.; Piscitelli, F.; Rosen, H.; et al. A Global Map of Lipid-Binding Proteins and Their Ligandability in Cells. Cell 2015, 161, 1668–1680.
- Hulce, J.J.; Cognetta, A.B.; Niphakis, M.J.; Tully, S.E.; Cravatt, B.F. Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat. Methods 2013, 10, 259–264.
- Kidd, D.; Liu, Y.; Cravatt, B.F. Profiling serine hydrolase activities in complex proteomes. Biochemistry 2001, 40, 4005–4015.
- Liu, Y.; Patricelli, M.P.; Cravatt, B.F. Activity-based protein profiling: The serine hydrolases. Proc. Natl. Acad. Sci. USA 1999, 96, 14694–14699.
- Kato, D.; Boatright, K.M.; Berger, A.B.; Nazif, T.; Blum, G.; Ryan, C.; Chehade, K.A.; Salvesen, G.S.; Bogyo, M. Activity-based probes that target diverse cysteine protease families. Nat. Chem. Biol. 2005, 1, 33–38.
- Hewings, D.S.; Heideker, J.; Ma, T.P.; AhYoung, A.P.; El Oualid, F.; Amore, A.; Costakes, G.T.; Kirchhofer, D.; Brasher, B.; Pillow, T.; et al. Reactive-site-centric chemoproteomics identifies a distinct class of deubiquitinase enzymes. Nat. Commun. 2018, 9, 1162.
- Ward, J.A.; McLellan, L.; Stockley, M.; Gibson, K.R.; Whitlock, G.A.; Knights, C.; Harrigan, J.A.; Jacq, X.; Tate, E.W. Quantitative Chemical Proteomic Profiling of Ubiquitin Specific Proteases in Intact Cancer Cells. ACS Chem. Biol. 2016, 11, 3268–3272.
- Zhao, Q.; Ouyang, X.; Wan, X.; Gajiwala, K.S.; Kath, J.C.; Jones, L.H.; Burlingame, A.L.; Taunton, J. Broad-Spectrum Kinase Profiling in Live Cells with Lysine-Targeted Sulfonyl Fluoride Probes. J. Am. Chem. Soc. 2017, 139, 680–685.
- Lebraud, H.; Wright, D.J.; East, C.E.; Holding, F.P.; O’Reilly, M.; Heightman, T.D. In-gel activity-based protein profiling of a clickable covalent ERK1/2 inhibitor. Mol. Biosyst. 2016, 12, 2867–2874.
- Tan, J.; Yang, X.; Zhuang, L.; Jiang, X.; Chen, W.; Lee, P.L.; Karuturi, R.K.; Tan, P.B.; Liu, E.T.; Yu, Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007, 21, 1050–1063.
- Chase, A.; Cross, N.C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618.
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29.
- Yang, P.Y.; Liu, K.; Ngai, M.H.; Lear, M.J.; Wenk, M.R.; Yao, S.Q. Activity-based proteome profiling of potential cellular targets of Orlistat-an FDA-approved drug with anti-tumor activities. J. Am. Chem. Soc. 2010, 132, 656–666.
- Shi, H.; Cheng, X.; Sze, S.K.; Yao, S.Q. Proteome profiling reveals potential cellular targets of staurosporine using a clickable cell-permeable probe. Chem. Commun. 2011, 47, 11306–11308.
- Shi, H.; Zhang, C.J.; Chen, G.Y.; Yao, S.Q. Cell-based proteome profiling of potential dasatinib targets by use of affinity-based probes. J. Am. Chem. Soc. 2012, 134, 3001–3014.
- Yang, P.Y.; Wang, M.; He, C.Y.; Yao, S.Q. Proteomic profiling and potential cellular target identification of K11777, a clinical cysteine protease inhibitor, in Trypanosoma brucei. Chem. Commun. 2012, 48, 835–837.
- Tam, E.K.; Li, Z.; Goh, Y.L.; Cheng, X.; Wong, S.Y.; Santhanakrishnan, S.; Chai, C.L.; Yao, S.Q. Cell-based proteome profiling using an affinity-based probe (AfBP) derived from 3-deazaneplanocin A (DzNep). Chem. Asian J. 2013, 8, 1818–1828.
- Prasch, J.; Bernhart, E.; Reicher, H.; Kollroser, M.; Rechberger, G.N.; Koyani, C.N.; Trummer, C.; Rech, L.; Rainer, P.P.; Hammer, A.; et al. Myeloperoxidase-Derived 2-Chlorohexadecanal Is Generated in Mouse Heart during Endotoxemia and Induces Modification of Distinct Cardiomyocyte Protein Subsets In Vitro. Int. J. Mol. Sci. 2020, 21, 9235.
- Ji, C.; Kozak, K.R.; Marnett, L.J. IkappaB kinase, a molecular target for inhibition by 4-hydroxy-2-nonenal. J. Biol. Chem. 2001, 276, 18223–18228.
- Neely, M.; Sidell, K.R.; Graham, D.G.; Montine, T.J. The Lipid Peroxidation Product 4-Hydroxynonenal Inhibits Neurite Outgrowth, Disrupts Neuronal Microtubules, and Modifies Cellular Tubulin. J. Neurochem. 2010, 72, 2323–2333.
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913.
- Levonen, A.L.; Landar, A.; Ramachandran, A.; Ceaser, E.K.; Darley-Usmar, V.M. Cellular mechanisms of redox cell signalling: Role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem. J. 2004, 378, 373–382.
- Vila, A.; Tallman, K.A.; Jacobs, A.T.; Liebler, D.C.; Porter, N.A.; Marnett, L.J. Identification of protein targets of 4-hydroxynonenal using click chemistry for ex vivo biotinylation of azido and alkynyl derivatives. Chem. Res. Toxicol. 2008, 21, 432–444.
- Cohen, M.S. Interplay between compartmentalized NAD+ synthesis and consumption: A focus on the PARP family. Genes Dev. 2020, 34, 254–262.
- Sileikyte, J.; Sundalam, S.; David, L.L.; Cohen, M.S. Chemical Proteomics Approach for Profiling the NAD Interactome. J. Am. Chem. Soc. 2021, 143, 6787–6791.
- Ravindran, M.S.; Wenk, M.R. Activity-Based Lipid Esterase Profiling of M. bovis BCG at Different Metabolic States Using Tetrahydrolipstatin (THL) as Bait. Methods Mol. Biol. 2017, 1491, 75–85.
- Adam, G.C.; Vanderwal, C.D.; Sorensen, E.J.; Cravatt, B.F. (-)-FR182877 is a potent and selective inhibitor of carboxylesterase-1. Angew. Chem. Int. Ed. Engl. 2003, 42, 5480–5484.
- Luo, Y.; Knuckley, B.; Bhatia, M.; Pellechia, P.J.; Thompson, P.R. Activity-based protein profiling reagents for protein arginine deiminase 4 (PAD4): Synthesis and in vitro evaluation of a fluorescently labeled probe. J. Am. Chem. Soc. 2006, 128, 14468–14469.
- Jones, J.E.; Causey, C.P.; Knuckley, B.; Slack-Noyes, J.L.; Thompson, P.R. Protein arginine deiminase 4 (PAD4): Current understanding and future therapeutic potential. Curr. Opin. Drug Discov. Dev. 2009, 12, 616–627.
- Vossenaar, E.R.; Zendman, A.J.; van Venrooij, W.J.; Pruijn, G.J. PAD, a growing family of citrullinating enzymes: Genes, features and involvement in disease. Bioessays 2003, 25, 1106–1118.
- Stone, E.M.; Schaller, T.H.; Bianchi, H.; Person, M.D.; Fast, W. Inactivation of Two Diverse Enzymes in the Amidinotransferase Superfamily by 2-Chloroacetamidine: Dimethylargininase and Peptidylarginine Deiminase. Biochemistry 2005, 44, 13744–13752.
- Bicker, K.L.; Thompson, P.R. The protein arginine deiminases: Structure, function, inhibition, and disease. Biopolymers 2013, 99, 155–163.
- Ghari, F.; Quirke, A.M.; Munro, S.; Kawalkowska, J.; Picaud, S.; Mcgouran, J.; Subramanian, V.; Muth, A.; Williams, R.; Kessler, B. Citrullination-acetylation interplay guides E2F-1 activity during the inflammatory response. Sci. Adv. 2016, 2, 1501257.
- Knight, J.S.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Smith, C.K.; Hodgin, J.B.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann. Rheum. Dis. 2015, 74, 2199–2206.
- Kawalkowska, J.; Quirke, A.M.; Ghari, F.; Davis, S.; Subramanian, V.; Thompson, P.R.; Williams, R.O.; Fischer, R.; La Thangue, N.B.; Venables, P.J. Abrogation of collagen-induced arthritis by a peptidyl arginine deiminase inhibitor is associated with modulation of T cell-mediated immune responses. Sci Rep. 2016, 6, 26430.
- Speers, A.E.; Adam, G.C.; Cravatt, B. Activity-Based Protein Profiling in Vivo Using a Copper(I)-Catalyzed Azide-Alkyne (3 + 2) Cycloaddition. J. Am. Chem. Soc. 2003, 125, 4686–4687.
- Chang, J.W.; Cognetta, A.B.; Niphakis, M.J.; Cravatt, B.F. Proteome-wide reactivity profiling identifies diverse carbamate chemotypes tuned for serine hydrolase inhibition. ACS Chem. Biol. 2013, 8, 1590–1599.
- Erlanson, D.A.; Braisted, A.C.; Raphael, D.R.; Randal, M.; Stroud, R.M.; Gordon, E.M.; Wells, J.A. Site-directed ligand discovery. Proc. Natl. Acad. Sci. USA 2000, 97, 9367–9372.
- Mamidyala, S.K.; Finn, M.G. In situ click chemistry: Probing the binding landscapes of biological molecules. Chem. Soc. Rev. 2010, 39, 1252–1261.
- Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discov. 2002, 1, 493–502.
- Brik, A.; Wu, C.Y.; Wong, C.H. Microtiter plate based chemistry and in situ screening: A useful approach for rapid inhibitor discovery. Org. Biomol. Chem. 2006, 4, 1446–1457.
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315.
- Kalesh, K.A.; Liu, K.; Yao, S.Q. Rapid synthesis of Abelson tyrosine kinase inhibitors using click chemistry. Org. Biomol. Chem. 2009, 7, 5129–5136.
- Klein, M.; Diner, P.; Dorin-Semblat, D.; Doerig, C.; Grotli, M. Synthesis of 3-(1,2,3-triazol-1-yl)- and 3-(1,2,3-triazol-4-yl)-substituted pyrazolo[3,4-d]pyrimidin-4-amines via click chemistry: Potential inhibitors of the Plasmodium falciparum PfPK7 protein kinase. Org. Biomol. Chem. 2009, 7, 3421–3429.
- Smith, W.L.; Urade, Y.; Jakobsson, P.J. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 2011, 111, 5821–5865.
- Bhardwaj, A.; Kaur, J.; Wuest, M.; Wuest, F. In situ click chemistry generation of cyclooxygenase-2 inhibitors. Nat. Commun. 2017, 8, 1–14.
- Torres, C.R.; Hart, G.W. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 1984, 259, 3308–3317.
- Wang, Y.; Zhu, J.; Zhang, L. Discovery of cell-permeable O-GlcNAc transferase inhibitors via tethering in situ click chemistry. J. Med. Chem. 2017, 60, 263–272.
- Hirsh, A.J.; Yao, S.Y.M.; Young, J.D.; Cheeseman, C.I. Inhibition of glucose absorption in the rat jejunum: A novel action of alpha-D-glucosidase inhibitors. Gastroenterology 1997, 113, 205–211.
- Yamagishi, S.-I.; Matsui, T.; Ueda, S.; Fukami, K.; Okuda, S. Clinical Utility of Acarbose, an α-Glucosidase Inhibitor in Cardiometabolic Disorders. Curr. Drug Metab. 2009, 10, 159–163.
- Hara, T.; Nakamura, J.; Koh, N.; Sakakibara, F.; Hotta, N. An importance of carbohydrate ingestion for the expression of the effect of alpha-glucosidase inhibitor in NIDDM. Diabetes Care 1996, 19, 642–647.
- Wang, G.; Peng, Z.; Wang, J.; Li, J.; Li, X. Synthesis and biological evaluation of novel 2,4,5-triarylimidazole–1,2,3-triazole derivatives via click chemistry as α-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 5719–5723.

