1. Activity-Based Protein Profiling
Over the past few years, activity-based protein profiling (ABPP) has become a strong method of chemical proteomics for analyzing proteins’ functional states within a complex proteome [
6,
7]. ABPP strategies usually use activity-based probes (ABPs), that are designed to be recognized by the target protein and react with residues from the active site, which can efficiently enrich and identify of low-abundance and low-affinity probe-interacting proteins [
8]. Many ABPs have been developed for many classes of enzymes, including serine hydrolases [
9,
10], cysteine proteases [
11], metallohydrolases, phosphatases, deubiquinating enzymes [
12,
13], kinases [
14,
15], various oxidoreductases, and others. Although ABPs often utilize reporter groups for the direct enrichment or visualization of labeled proteins, avoiding the need for additional conjugation steps, such bulky groups can hamper cellular uptake and tissue distribution, potentially limiting their application in living systems. To improve these problems, compound-centric chemical proteomics approach on the basis of click reactions were used to enrich and identify enzyme targets, which allow for the incorporation of chemical groups with highly selective reactivity into small molecules, or protein modifications without perturbing their biological function, enabling the selective installation of an analysis tag for downstream investigations (
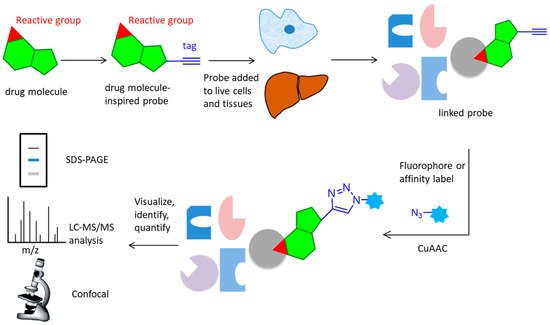
Figure 1). The probe is designed based on the structure of the active molecule, then added to live cells or tissues. It reacts covalently (via an electrophilic trap or a photo-crosslinking group) or non-covalently with the target protein. The lysed samples subject to CuAAC reaction attach a fluorophore, affinity label, or a combination of these elements. Marked proteins are subsequently visualized, identified or quantified using a variety of techniques, such as SDS-PAGE, LC-MS/MS analysis or confocal imaging [
8]. The CuAAC-enabled ABPs were developed for protein arginine deiminases [
16]; ubiquitin mechanisms [
12,
17], cytochrome P450 enzymes [
18], glycosidases [
19], and kinase [
20,
21].
Figure 1. Overall workflow of the activity-based protein profiling approach. A probe with an active group and an alkynyl tag reacts with the active site of target protein in live cells and tissues, then undergoes CuAAC reaction with an azide containing fluorophore or affinity label. After lysis and separation, the probe is fluorescently scanned in SDS-PAGE display, LC-MS/MS analysis, or confocal imaging for identification or quantification.
Clickable probes based on light affinity can be the most common chemical proteomics tool for capturing and identifying non-covalent targets for small bioactive molecules [
22]. These probes include the synthesis and installation of a click group (azide or alkine) and photoaffinity groups (such as diazirines, benzophenone) to cause as little disruption as possible to the biological activity of the compounds, which sometimes necessitates a thorough SAR investigation and even new synthetic channels [
23,
24,
25]. Cell permeable sensors with both clickable group and light affinity have been developed to characterize a variety of clinically approved medications and inhibitors, including kinase inhibitors [
26], γ-secretory enzyme inhibitors [
27], β-secretor inhibitors [
28], antibiotics [
29], NSAIDs [
30], epigenetic regulatory compounds [
31], natural products [
32], and also protein interactions with lipids [
33] and sterol [
34]. Considering that metabolites may participate in functional sites of proteins, the metabolite-derived click probes can be used as a valuable analytical tool to plot drug interactions in the proteomes, and even as a tool for uncovering functional regulators of metabolite binding proteins [
33].
As a carbocyclic analog of adenosine, 3-Deazaneplanocin A (DzNep) can inhibit the activity of histone lysin methyltransferase, which has aroused great interest in epigenetic research over the past few years [
35,
36,
37]. However, the molecular mechanism and extracellular targets of DzNep have not been fully understood. Yang et al. developed some small-molecular probes derived from DzNep that are permeable to the cells, but the bulky modification groups in the probes usually disrupt the interactions between proteins and drugs, so it is still challenging to efficiently capture cell targets in situ interactions [
26,
38,
39,
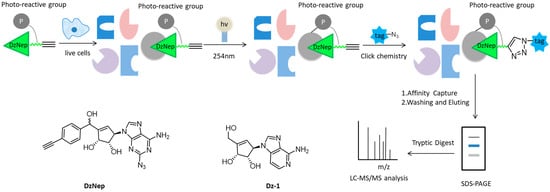
40]. Therewith, Tam and colleagues designed a novel “clickable” affinity-based probe DZ-1, with minimal structural modification from DzNep (
Figure 2). DZ-1 possessed comparative anti-apoptotic activity as DzNep in MCF-7 mammalian cells. In situ proteome profiling of DZ-1 was successfully carried out on the basis of pull-down LC-MS/MS analysis. Finally, some highly enriched proteins were identified as potential cellular protein targets of DzNep [
41].
Figure 2. Overall workflow of the cell-based proteome profiling approach followed by large-scale pull-down/LC-MS/MS for identification of potential cellular targets of DzNep using affinity-based probe DZ-1.
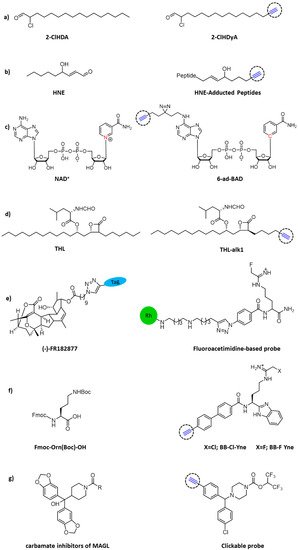
Inflammation-related processes are pivotal factors contributing to sepsis-associated cardiac dysfunction. Cardiac neutrophil infiltration and subsequent release of myeloperoxidase leads to the formation of the oxidant hypochlorous acid (HOCl) that is able to chemically modify plasmalogens into 2-chlorohexadecanal (2-ClHDA). To elucidate this metabolic process and characterize protein targets for 2-ClHDA, a clickable alkynyl analog, 2-chlorohexadec-15-in-1-al (2-ClHDyA), was used by Prasch and colleagues to identify its protein targets (
Figure 3a). Through CuAAC reaction of 5-tetramethylrhodamine azide (N3-TAMRA) and two-dimensional gel electrophoresis, they were able to pinpoint 51 proteins which form adducts with 2-ClhdYA. Genetic ontology enrichment analysis showed that heat shock and chaperone proteins, energetic metabolism and cytoskeleton proteins were the key targets for HOCl modified lipids in the heart of mice with endotoxemia [
42].
Figure 3. Chemical structures of clickable probes in ABPP for identifying protein targets of: (a) 2-ClHDA, (b) HNE, (c) NAD+, (d) THL, (e) natural product (-)–FR182877, (f) Cl- and F-amidine, and (g) carbamates.
Diffusible electrophilic α, β-unsaturated aldehydes, such as 4-hydroxynonenal (HNE), are primary targets of free radical damage during oxidative stress. Some studies have shown that HNE or other electrophilic agents can modify IκB kinase (IKK) [
43], tubulin isomer [
44], and Keap1 [
45,
46], leading to the loss of protein function and disturbance of cell signal transmission. In order to fully understand the effect of oxidative stress on cellular signaling transduction and disease pathology, it is necessary to analyze HNE modified proteins in vivo. Vila and colleagues explored Staudinger’s ligation and CuAAC to selectively label proteins with HNE in colon cancer cells, and subsequently pull-down by biotin–streptavidin interaction for LC-MS/MS analysis (
Figure 3b). The results showed that both strategies produced effective biotinylation of HNE-conjugated protein, while click chemistry was proven to be superior for recovering proteins [
47].
Nicotinamide adenine dinucleotide (NAD
+), known as oxidoreductase coenzyme, is also a multifunctional substrate of many post-translational modification enzymes, such as poly-ADP−ribose polymerases (PARP) and sirtuins [
48]. The recent studies of noncanonical NAD-binding proteins suggest that powerful chemical tools for profiling the NAD interactome are quite necessary. Šileikytė and colleagues developed a NAD
+/NADH probe, 6-ad-BAD, with two reactive sites for both click reaction and light crosslinking. Moreover, the nicotinamide linked to ribose was replaced by a benzamide adenine dinucleotide (BAD) to avoid enzyme digestion (
Figure 3c). Results showed that 6-ad-BAD could label PARP effectively in a UV dependent manner. Then, the chemical proteomics of 6-ad-BAD was evaluated in HEK 293T cell lysate through biotinylated enrichment and 24 unknown NAD or related nucleotide binding proteins were identified. This clickable probe will be useful in future chemical proteomics studies for profiling the NAD
+ interactome across different tissues as well as in disease contexts [
49].
Tetrahydrolipstatin (THL, also known as Orlistat) is an FDA approved anti-obesity drug with potential bactericidal activity. To explore the enzymatic targets of this β-lactam ring in a complex bacterial proteome, Ravindran and Wenk designed a functional THL–alkyne analog (
Figure 3d) to quantify the lipid esterase activity and enriched the target proteins in
Mycobacterium bovis BCG at different physiologic states [
50].
Adam and colleagues used click chemistry as a handy binding method to synthesize both rhodamine-, and rhodamine and biotin-tagged (trifunctional) sensors from the natural product (-)–FR182877 [
51]. Using this sensor, the researchers identified carboxyesterase-1 as the protein target in the heart of the mouse (
Figure 3e). In the same way, Thompson et al. used click chemistry to covalently label rhodamine with inhibitors for arginine deiminase 4 (PAD4), an enzyme controlled by calcium [
52].
Citrullination is the post-translational hydrolysis of peptidyl-arginine to form peptidyl-citrulline [
53,
54,
55], which is a reaction also catalyzed by PADs. Abnormal increase of citrullinated protein is associated with autoimmune illnesses and cancers [
53,
56]. Cl-amidine and F-amidine were reported to permanently inhibit PADs by covalently altering an active cysteine site [
57,
58,
59]. Herein, Nemmara and colleagues developed cell permeable and “clickable” probes (BB-Cl-Yne and BB-F-Yne) for covalent labeling of the PADs both in vitro and in cell-based systems [
9]. These sensors covalently alter the conserved cysteine residues in all PAD isozymes and serve as the base for azide-alkyne cycloaddition, and, subsequently, CuAAC [
60] with either TAMRA-N
3 or biotin-N
3. It is worth noting that such compounds may be used in several forms, including the off-target recognition of the parent compounds and ABPPs on the target binding tests, to demonstrate PAD inhibitor efficacy.
Chang and colleagues used a combination of competitive and click chemistry ABPP (
Figure 3g) to study a variety of proteomic reactions to activate carbamates in vitro and in vivo. They identified several carbamates derivatives, among them,
O-aryl and
O-hexafluoroisopropyl (HFIP) carbamates could react selectively with serum hydrolases in vivo. They used the proteomic specificity of carbamate HFIP to plot in situ images of monoacylglycerol lipase endocannabinoid hydrolases and
α-β hydrolase-6. They proved that carbamates are the preferred reaction group of serine hydrolase, which can adapt to different structural modifications and produce inhibitors with special potency and selectivity in the mammalian proteome [
61].
Designed to identify chemical probes for functionally related protein families that can be used in complex natural environments, ABPP is an area that benefits from click chemistry. Due to the modular and efficient properties of the click reaction, the synthesis of probe libraries has been greatly simplified. However, there is still lack of a universal chemical probe or small molecule ligand for all target proteins, and the real-time dynamic imaging of a specific protein of interest in a live organism remains highly challenging. Due to the diversity of biological applications, there is no standardized protocol covering the different aspects of probe preparation, click reaction conditions, or analysis. This results in various methods that may seem overwhelming to the newcomer. Click reactions can be readily integrated to both conventional biological techniques such as gel-based fluorescent-labeling, biotin-based pull-down assay, and many of the upcoming high-throughput bioassays and characterization techniques such as microarray, LC-MS/MS, etc. Furthermore, click modules enable the enrichment of protein targets, yet their presence can sometimes perturb molecular interactions and biological activity. Recent advances in ‘‘label-free’’ proteomic methods, such as thermal proteome profiling or drug affinity responsive target stability, represent complementary strategies to plot small molecule–protein interactions, bypassing the requirement of enrichment handles.