Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dimitrios Goutas | + 3735 word(s) | 3735 | 2021-09-23 05:29:15 | | | |
| 2 | Bruce Ren | -13 word(s) | 3722 | 2021-09-24 03:12:43 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Goutas, D. HuR in Pancreatic Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/14488 (accessed on 05 July 2026).
Goutas D. HuR in Pancreatic Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/14488. Accessed July 05, 2026.
Goutas, Dimitrios. "HuR in Pancreatic Cancer" Encyclopedia, https://encyclopedia.pub/entry/14488 (accessed July 05, 2026).
Goutas, D. (2021, September 23). HuR in Pancreatic Cancer. In Encyclopedia. https://encyclopedia.pub/entry/14488
Goutas, Dimitrios. "HuR in Pancreatic Cancer." Encyclopedia. Web. 23 September, 2021.
Copy Citation
Pancreatic cancer is set to become the most lethal and common type of cancer worldwide. This is partly attributed to the mutational burden that affects core signaling pathways and the crosstalk of tumor cells with their surrounding microenvironment, but it is also due to modern eating habits. Hyperadiposity along with the constant rise in metabolic syndrome’s incidence contribute to a state of metaflammation that impacts immune cells and causes them to shift towards an immunosuppressive phenotype that, ultimately, allows tumor cells to evade immune control.
HuR
pancreatic adenocarcinoma
pathogenesis
treatment
chemoresistance
tumor microenvironment
1. Introduction
Pancreatic ductal adenocarcinoma (PDA) represents the fourth (4th) most common cause of cancer mortality in developed countries [1], with geographical variations and lifestyle factors shaping the context of its incidence. Contrary to the other cancer entities, pancreatic cancer has been linked to many risk factors involved in several different pathways, including hereditary and genetic factors [1]. Unfortunately, the vast majority of pancreatic adenocarcinomas have already spread beyond the pancreatic parenchyma at the time of diagnosis, mostly extending into the ampulla of Vater, the duodenum and the intrapancreatic portion of the common bile duct, as well as into the peripancreatic or retroperitoneal adipose tissue [2]. Several tumor suppressor genes have been linked to pancreatic neoplasia, such as Kirsten rat sarcoma (KRAS), cyclin dependent kinase 4A (CDK4A), tumor suppressor protein 53 (TP53) and the SMAD family member 4 (SMAD4) [3][4]. More commonly, SMAD4 seems to be inactivated in 55% of pancreatic cancers, either by homozygous deletion or by loss of one allele coupled with an intragenic mutation in the second allele [5]. These mutations lead to the dysregulation of core signaling pathways, affecting both the proliferation and migration of tumor cells and also the crosstalk with their surrounding tumor microenvironment (TME) [6]. A large genomic analysis study performed by Bailey et al. [7] in 2016 of 456 PDAs led to the identification of 32 recurrently mutated genes that ultimately resulted in classifying these tumors into four subtypes, each of them correlated with specific histopathologic characteristics. Namely, these subtypes include (a) squamous; (b) pancreatic progenitor; (c) immunogenic and (d) aberrantly differentiated endocrine exocrine (ADEX) [7]. Based on the World Health of Organization (WHO), PDA is classified according to histology into colloid carcinoma (CC), signet-ring cell carcinoma (SRCC), undifferentiated carcinoma with osteoclast-like giant cells (UCOGC), adenosquamous carcinoma (ASC) and hepatoid, rhabdoid and medullary carcinoma [8]. To date, PDA prognosis remains dismal, with an average survival of 3–5 months for untreated patients and 10–20 months for patients undergoing surgical resection [9][10]; adjuvant chemotherapy with gemcitabine or 5-fluorouracil (5-FU) prolongs survival only slightly [11]. The failure of “all-comer” treatment approach, along with the molecular diversity and the unique tumor microenvironment of PDA [12][13][14], makes the turn towards precision medicine more paramount than ever. Advances in genomic-driven personalized medicine have shown promise in identifying unique therapeutic targets in individual patients, personalizing treatment selection. However, the ability of PDA cancer cells to compensate through different signaling pathways, as well as the fact that various genetic lesions are found to be pivotal for tumor progression, constitutes a challenge for this approach. HuR, an RNA binding protein, participates in posttranscriptional control of RNAs, such as splicing, polyadenylation, mRNA stabilization, localization and translation [15]. Additionally, the HuR molecule controls gene expression in multiple areas of malignant transformation, thus regulating the expression of multiple cancer-related genes. Its protein levels and localization have been linked to pathologic inflammation, malignant transformation and cancer progression by evading immune destruction, inducing angiogenesis and promoting tumor-associated inflammation [16][17][18].
Taking the above into consideration, one should consider a target that can be stimulated by the unique tumor microenvironment of PDA cancer cells (Figure 1), a target that can be found in abundance in cancer cells, while not in pancreatic normal cells, and a target that can offer a significant survival advantage in PDA cancer cells. In this review, we focus on HuR, an RNA-binding protein, and its therapeutic significance in pancreatic neoplasia and inflammation.
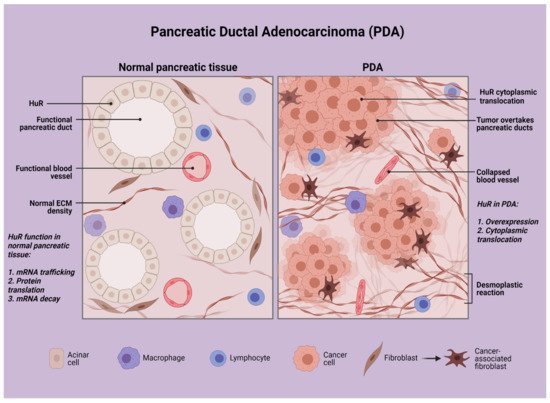
Figure 1. Schematic representation of pancreatic ductal adenocarcinoma in contrast with normal pancreatic tissue. Created with BioRender.
2. HuR and Neoplasia
HuR is an RBP encoded by the ELAVL1 gene that acts by stabilizing mRNAs and regulating gene expression [19]. HuR is composed of various structural motifs, such as RNA recognition motif (RRM), dsRNA binding domain, zinc fingers and others. RBPs have crucial roles in various cellular processes, such as cellular function, transport and localization. Posttranscriptional control of RNAs, such as splicing, polyadenylation, mRNA stabilization, localization and translation are some of the major functions displayed by RBPs [15]. HuR encodes a 32 kD protein composed of three RNA-binding domains that belong to the RRM family. More specifically, it is RRM-1 and RRM-2 that are responsible for AU rich elements (ARE) binding, and it is RRM-3 that binds to the mRNA poly(A) tail [20]. The expression of various molecules is regulated by HuR protein through different posttranscriptional mechanisms, including mRNA trafficking, protein translation and mRNA decay. Furthermore, HuR can control gene expression in multiple areas of malignant transformation, regulating multiple cancer related genes’ expression. HuR overexpression has been repeatedly linked to malignant transformation, and increased nuclear/cytoplasmic HuR expression has been associated with patients’ prognosis in different malignancies [21]. The above-mentioned properties advocate for the adverse clinical outcomes related to HuR protein overexpression in various cancer types, including advanced stage, positive lymph nodes and poor survival [22][23][24][25][26][27].
It has been demonstrated that posttranscriptional gene regulation allows PDA and colorectal tumor cells to survive under cancer-associated stress conditions [28][29][30][31][32][33]. More specifically, RBPs and micro-RNAs (miRNAs) contribute to posttranscriptional gene regulation and can directly influence gene expression by modulating mRNA stability [34][35]. Among the many RBPs and miRNAs that repress translation and foster mRNA decay, HuR represents the most potent and promising agent against cancer-associated mRNA degradation. Constantino et al. [29] studied the consequences of adjusting HuR levels in pancreatic cancer cells, and they found out that the cells overexpressing HuR in their cytoplasm were highly more sensitive to gemcitabine. More specifically, in pancreatic cancer cells, deoxycytidine kinase (dCK) mRNA interacts with HuR encoding the enzyme that metabolizes and activates gemcitabine [29]. Gemcitabine activation enhances the association of HuR with dCK mRNA and consequently increases HuR’s cytoplasmic expression. Appropriately, HuR’s overexpression leads to dCK mRNA increase in pancreatic cancer cells, while its silencing reduces dCK levels. Additionally, a clinical trial performed by Constantino et al. on gemcitabine efficacy demonstrated that patients with low HuR cytoplasmic expression levels had a 7-fold increased mortality risk when compared with patients with elevated HuR cytoplasmic levels [29]. Their results enhance the belief of HuRs’ paramount role as a key mediator of gemcitabine efficacy in PDA through the posttranscriptional regulation of dCK mRNA levels.
Additionally, the harsh TME of PDAs favors the growth of the most aggressive and suitable PDA cells. As PDA tumors are set in a hypoxic, nutrient-deprived microenvironment, only the most appropriate and aggressive clonal populations will survive and thrive [36][37]. These populations are also the most resistant against the cytotoxic chemotherapeutic agents used in PDAs [4][37][38]. Consequently, PDA cells, in order to survive under these hypoxic conditions, put together a multifaceted response and activate hypoxia inducible factors (HIF), such as the proviral integration site for Moloney murine leukemia virus 1 (PIM1) and HuR [39][40]. Furthermore, HuR protects PDA cells under nutrient-deprived conditions by regulating the key metabolic enzyme isocitrate dehydrogenase 1 (IDH1) [41] and translocating it into the cytoplasm [42]. It is via the regulation of these cellular reprogramming events that HuR activates various pathways for regulating angiogenesis, intracellular pH, DNA repair, cell survival, cell motility and mitochondrial function [42][43][44].
3. Elevated HuR Expression Causes a Pancreatitis-Like Inflammatory Microenvironment
The role of local and systemic inflammation has been shown to be imperishably linked to PDA growth, development and metastasis [45]. The inflammatory and cancer cells of a tumor’s microenvironment are capable of producing and releasing a variety of anti- and proinflammatory cytokines and regulating the balance among them through their constant interactions [46]. The cytokines more commonly implicated in pancreatic cancer are the anti-inflammatory cytokines tumor growth factor β (TGF-β) and interleukin (IL)-10 and the pro-inflammatory IL-6 and IL-1β. IL10 and tumor necrosis factor-α (TNF-α) play a paramount role in the microenvironment of PDA tumors [46]. Low-grade chronic inflammation in systemic diseases, such as metaflammation in patients with metabolic syndrome or diabetes mellitus (DM), enhance the risk of cancer, particularly pancreatic cancer [47]. This subclinical chronic inflammation linked with hyperadiposity or DM enhances the belief that there is a link between chronic inflammation and pancreatic cancer [47]. More specifically, obese patients demonstrate an augmented release of the pro-inflammatory adipokine leptin and a reduced release of the anti-inflammatory adipokine adiponectin, while at the same time, a shift from the M2 to the M1 macrophages infiltrating the adipose tissue triggers the release of pro-inflammatory cytokines, predominately TNF-α and IL-6 [48]. Nevertheless, in addition to the role of metaflammation in PDA in patients with metabolic syndrome, the consequences of pathways linked to nutrition and the gut microbiome also seem to contribute significantly to pancreatic neoplasia [49][50]. Excessive eating exerts a solid immunomodulatory effect by giving rise to a subclinical inflammatory status with pro-inflammatory cytokines and trophic hormones and by impacting immune cells through the gut microbiota, which can ultimately lead to a shift towards an immunosuppressive phenotype that allows tumor cells to escape immune control [48].
HuR’s role in inflammation is now long established [21][51]. HuR regulates mRNAs responsible for encoding proinflammatory proteins (TNF-α, IL-6, COX-2) but also proteins that block the production of anti-inflammatory factors (thrombomodulin). More specifically, HuR has been implicated in several diseases for augmenting inflammation, including asthma, inflammatory bowel disease and rheumatoid arthritis, among others [52]. Peng et al. [16] studied the consequences of HuR overexpression in a transgenic mouse model that had a >2-fold elevation of pancreatic HuR expression. Histological examination revealed an intense fibroinflammatory reaction, along with a marked increase in inflammatory infiltrates, fibrosis, ductal complexes and acinar atrophy—features suggestive of chronic pancreatitis [16]. Moreover, immunohistochemical examination showed an increased expression of TNFα, cyclooxygenase-2 (COX-2), vimentin, α-smooth muscle actin (α-SMA) and collagen-1 [16]. Additionally, cluster of differentiation (CD) [45], CD3, CD86 and IL-6 were increased in the transgenic mouse model in comparison with the mice without HuR overexpression, further outlining the fibroinflammatory response commenced by HuR protein. Additionally, Peng et al. attempted to correlate HuR’s expression with tumorigenesis by examining hematoxylin and eosin (H/E) sections from pancreata of TC mice older than 10 months old, without, however, any precancerous pancreatic intraepithelial neoplasia (PanIN) lesions or PDA being observed; this led to the conclusion that in the absence of any known driver of gene mutations, HuR overexpression alone does not initiate tumorigenesis [16]. Nevertheless, when combining KRAS mutant mice with HuR overexpression compared with only KRAS mutant mice, they observed an increased incidence of PanIN lesions and up to 5-fold increased incidence of PDAs in the mice overexpressing HuR, supporting the notion that the inflammatory microenvironment induced by HuR expression in KRAS mutant mice promotes tumor formation [16]. These findings directly incriminate HuR as a promoter of pancreatic cancer, predominantly within the context of inflammation.
4. Agents Interacting with HuR Expression and How HuR’s Inhibition Could Affect Tumor Progression
Many different strategies have been used in an attempt to modify or suppress HuR’s action in cancer [27], including inhibiting its cytoplasmic translocation, decreasing its expression via siRNAs or inhibiting its binding to target mRNAs. On top of that, intense research has been performed to obtain data regarding the synergistic use of HuR inhibition with chemotherapeutic and a variety of other agents [53][54][55][56][57] (Figure 2, Table 1).
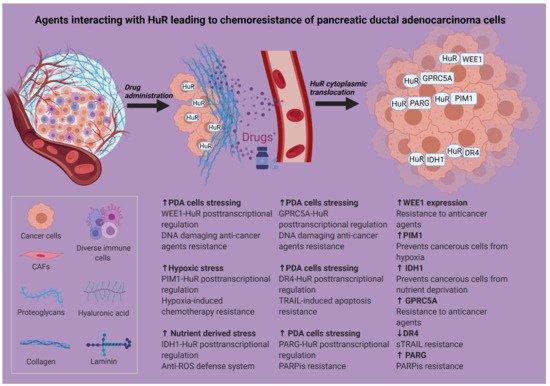
Figure 2. Agents interacting with HuR causing chemoresistance of pancreatic ductal adenocarcinoma cells. Created with BioRender. Abbreviations in Figure 2: CAFs: cancer-associated fibroblasts; DR4: death receptor 4; ECM: extracellular matrix; GPRC5A: G protein-coupled receptor class C group 5 member A; HuR: human antigen R; IDH1: isocitrate dehydrogenase; PARG: poly(ADP-ribose) glycohydrolase; PARP: poly (ADP-ribose) polymerase; PARPis: poly (ADP-ribose) polymerase inhibitors; PIM1: proviral integration site for Moloney murine leukemia virus-1; ROS: reactive oxygen species; sTRAIL: soluble TRAIL; TRAIL: TNF-related apoptosis-inducing ligand; HuR: human antigen R. Created with BioRender. * Gemcitabine, 5-fluorouracil, oxaliplatin.
Table 1. Agents interacting with HuR in a posttranscriptional level and their therapeutic outcome.
| Agent | Function | Expression | HuR Action | Mechanism | Outcome | Ref. |
|---|---|---|---|---|---|---|
| PIM1 | Serine-threonine kinase | ↑ | Cytoplasmic translocation Binds PIM1 mRNA |
Phosphorylation and inactivation of BAD Activation of MEK 1/2 |
Prevents cancerous cells from hypoxia | [41] |
| IDH1 | NADPH generating enzyme | ↑ | Cytoplasmic translocation Binds IDH1 mRNA |
HuR impacts antioxidant defense by regulating IDH1 | Prevents cancerous cells from nutrient deprivation | [42] |
| WEE1 | Mitotic inhibitor kinase | ↑ | Cytoplasmic translocation Interacts with WEE1 mRNA |
CDK1-phosphorylation Increase in the γH2AX levels Cell cycle arrest at the G2/M transition |
Resistance to anti-cancer agents | [32] |
| Abemaciclib | Chemotherapeutic agent | --- | HuR inhibition | CMLD-2 and pyrvinium pamoate | Decreased IC50 rates to abemaciclib Increased sensitivity of PDA cells to abemaciclib |
[53] |
| GPRC5A | Protein binding gene Dual behavior (oncogene or tumor suppressor) |
↑ | HuR cytoplasmic translocation Binds GPRC5A mRNA |
Posttranscriptional regulation | Monotonous increase in GPRC5A protein levels | [54] |
| TRAIL | Type II transmembrane protein | ↓ | HuR cytoplasmic translocation Binds DR4 mRNA |
Posttranscriptional regulation | Downregulation of TRAIL-induced DR4 mRNA expression Suppresses apoptosis |
[55] |
| PARPis | Inhibitors of PARP (family of proteins involved in several cellular processes) | --- | Cytoplasmic translocation Binds PARG mRNA |
HuR dependent stabilization of PARG | PARPis resistance | [57] |
4.1. Synergistic Use of HuR Inhibitors with Abemaciclib
Dhir et al. [53] analyzed the effect of combining abemaciclib with HuR inhibition by using two validated inhibitors, CMLD-2 and pyrvinium pamoate, in PDA cell lines [53]. The result was that both cell lines demonstrated a decreased number of colonies compared with monotherapy. Furthermore, PDA cell lines transfected with siHuR oligonucleotides, revealed decreased IC50 rates to abemaciclib in comparison with si-negative cells. This highlights the increased sensitivity that PDA cell lines obtained from the mentioned therapeutic combination regimen and the potential that this combination entails.
4.2. Agents Interacting with HuR Inducing Chemoresistance
PIM1 represents a hypoxia-inducing, pro-oncogenic, serine-threonine kinase that only recently turned out to be a key regulator of hypoxia-induced chemotherapy resistance in PDAs. The molecular mechanism underlying its overexpression in pancreatic carcinomas is based on the presence of cis-acting AREs in the PIM1’s mRNA 3′ untranslated region, which mediates an interaction with HuR in a tumor hypoxia context [41]. More specifically, HuR, in response to hypoxic stress, translocates from the nucleus to the cytoplasm of PDA cells and stabilizes PIM1 mRNA transcript, causing PIM1 protein overexpression. The HuR-mediated PIM1 protein overexpression prevents cancerous cells from hypoxia through phosphorylation and inactivation of BAD (Bcl-2-associated death promoter) and activation of MEK 1/2 (mitogen activated protein kinase kinase) [41]. Selective inhibition of HuR by MS-444 blocks its homodimerization and its cytoplasmic translocation, therefore rendering the PDA cells susceptible to oxaliplatin and 5-FU [41]. These results elucidate the role of HuR and its prosurvival properties in PDA and provide evidence that its selective inhibition and disruption of PIM1 regulation could be the key to interrupting this chemotherapeutic resistance mechanism.
IDH1 is a NADPH (nicotinamide adenine dinucleotide phosphate)-generating enzyme that has been demonstrated to be posttranscriptionally stabilized by HuR; via this posttranscriptional regulation, HuR manages an anti-ROS (reactive oxygen species) defense system. It is well known that HuR protects PDA cells not just from hypoxia but also from nutrient-derived stress [39][41]. A study conducted by Zarei et al. showed that, under nutrient-deprived conditions, PDA cells were less sensitive to gemcitabine in PDA xenografts in hypoglycemic mice, compared with the hyperglycemic mice [42]. Similar results were observed in a retrospective study of patients with elevated serum glucose levels treated with gemcitabine, as they revealed an improved OS (overall survival). Furthermore, Zarei et al. [42] identified an enhanced antioxidant defense as a driver of chemoresistance. More precisely, ROS levels were increased in vitro either by nutrient deprivation or gemcitabine treatment, but withdrawing nutrients from PDA cells before gemcitabine treatment enhanced this effect [42]. However, HuR expression reduced ROS levels under low glucose, whereas HuR silencing augmented ROS levels. Investigation via CRISPR and RNAi (RNA interference) of the factor responsible for maintaining survival of PDA cancer cells under nutrient-deprived conditions revealed HuR to be the implicated agent. Importantly, studies in HuR-null PDA cell lines demonstrated IDH1 as the single HuR-regulated antioxidant enzyme [42]. These findings support the notion that selective inhibition of HuR could break the HuR-IDH1 regulatory axis and serve as a promising therapeutic target.
Lal et al. [32] demonstrated that stressing PDA cancer cells with DNA damaging anti-cancer agents (carboplatin, cisplatin, oxaliplatin, mitomycin C and PARP-inhibitors) resulted in HuR’s translocation from the nucleus to the cytoplasm [32]. What is even more interesting, is that HuR knockdown in PDA cells resulted in their sensitization to the above agents, while HuR’s overexpression led to resistance. HuR was implicated with DNA-damaging anti-cancer agents by the acute upregulation of WEE1. Actually, WEE1, a mitotic inhibitor kinase, participates in the regulation of the DNA damage repair pathway, and its therapeutic inhibition along with chemotherapy is currently under clinical trials investigation for cancer treatment [58][59][60][61][62][63][64][65]. Furthermore, Lal et al. demonstrated the role of WEE1 as a HuR target, both in vitro and in vivo, by revealing the direct binding of HuR to WEE1 mRNA and that HuR small interfering RNA (siRNA) knockdown and/or overexpression affects the WEE1 protein levels, especially following DNA damage. HuR stimulation of WEE1 subsequently leads to an increase in the γH2AX levels, causes Cdk1 phosphorylation and facilitates cell cycle arrest at the G2/M transition. Additionally, they demonstrated a novel acute checkpoint mechanism that involves WEE1 and by which cells can block and potentially withstand any sudden DNA damage insult experienced [32]. Taking these parameters into consideration, targeting the HuR-WEE1 interactions could be a promising novel approach towards patients receiving chemotherapy and, thereby, enhancing their therapy’s outcomes. Generally, therapies focusing on translocating targets, such as HuR, and its targeting mRNAs, such as WEE1, could turn out to be more efficient therapeutic strategies than the targeting of PDA cells’ genetic alterations [32].
GPRC5A (G protein-coupled receptor class C group 5 member A) is a protein coding gene that demonstrates a dual behavior—acting as an oncogene in certain cancers and as a tumor suppressor in other cancers [66]. Zhou et al. [54] attempted to establish the impact of GPRC5A overexpression in PDA cell lines and provided an association between its overexpression and HuR’s role in pancreatic cancer. After exhibiting that GPRC5A mRNA levels hold the second highest average expression among different cancer types in pancreatic cancer, they examined and compared its expression levels in normal pancreatic tissues, primary PDAs and metastatic tumors. As a result, GPRC5A was shown to be overexpressed in primary and metastatic PDA tumors, and even more in the metastatic sites [54]. This was further validated through immunohistochemical analysis. In an attempt to analyze the posttranscriptional regulation of GPRC5A, the authors performed luciferase assays, demonstrating that HuR binds to at least one site in the 3′-UTR of GPRC5A. More specifically, after the cellular stress caused by gemcitabine treatment, HuR translocates to the cytoplasm where it binds with the GPRC5A mRNA and induces a monotonous increase in GPRC5A protein levels for at least 18 h. After this, HuR and GPRC5A mRNA association returns to background levels and its posttranscriptional control decreases [54]. Additionally, GPRC5A levels were increased after treatment both with 5-FU and oxaliplatin suggesting that other factors could also be involved in regulating GPRC5A expression in response to chemical stressors. These interactions of GPRC5A with HuR, gemcitabine and the other chemotherapeutic agents imply a potential pro-oncogenic role for this gene; therefore, targeting of these interactions could augment the death rate of pancreatic cancer cells post-chemotherapy treatment.
TRAIL (TNF-related apoptosis-inducing ligand) is a type II transmembrane protein harvesting an important role in cancer onset, progression and apoptosis [67]. TRAIL directly induces apoptosis by engaging cell surface death receptors (DR) DR4 and DR5, constituting a possible molecular target in cancer therapeutics. It has been previously demonstrated that HuR and DR5 expression share an inverse relation in vitro and in PDA patient tissues. Additionally, HuR is capable of binding to DR5 mRNA and suppressing its protein expression, leading to a decrease in apoptosis [68]. Nevertheless, due to the fact that DR4, and not DR5, has been proven to be a more potent trigger for TRAIL-induced apoptosis in PDA cells [69], Romeo et al. studied the effects of HuR levels and their correlation with DR4 expression levels and TRAIL resistance in PDA [55]. In order to obtain information regarding the association of DR4 and DR5 expression with PDA cell sensitivity to soluble (s)TRAIL treatment, the authors compared the IC50 for every cell line from the sTRAIL killing curve, revealing a strong direct correlation among DR4 cell surface expression and TRAIL sensitivity, compared with the association observed among DR5 expression and TRAIL sensitivity [55]. They further demonstrated that HuR not only translocates to the cytoplasm in response to sTRAIL treatment but also that HuR has the ability to posttranscriptionally bind DR4 mRNA through the 3′-UTR. Additionally, they utilized specific siRNA to silence HuR in PDA cell lines in the presence of sTRAIL, which resulted in an increase in DR4 cell surface protein expression, suggesting that HuR plays a role in the downregulation of TRAIL-induced DR4 mRNA expression [55]. Therefore, strategies focused on decreasing HuR cytoplasmic concentration in PDA patients could enhance the efficacy of certain treatment regimens, such as TRAIL.
Poly (ADP-ribose) polymerase (PARP) is a family of proteins involved in a number of cellular processes, such as DNA repair, genomic stability and programmed cell death [70]. Although PARP inhibitors (PARPi) initially showed promising results, it turned out that most tumors would develop drug resistance [71][72]. Chand et al. [73] demonstrated that the antitumor response to PARPi in PDA is largely controlled by the HuR-dependent stabilization of poly (ADP-ribose) glycohydrolase (PARG) [73]. More specifically, they attempted to assess the role of HuR in PARPi response in PDA cell lines via HuR’s knockout. As a result, CRISPR knockout of HuR ensued a 20-fold increase in sensitivity to several PARPis (olaparib and veliparib) [73]. Furthermore, they demonstrated that PARPis induced a cytoplasmic HuR translocation, which, however, could be blocked using a small molecule inhibitor, MS-444, that would prevent HuR dimerization. Additionally, HuR inhibition with MS-444 resulted in a significant decrease in PARG expression and an associated accumulation of total polyADP-ribosylation (PARylation) [73]. Taking these data together, inhibition of HuR inhibits PARG overexpression and function and could possibly be utilized to enhance the efficacy of PARPi.
Considering, the aforementioned, properties of HuR and the effects that it is capable of exerting both in tumor progression and in therapy induction, it automatically raises the probability of becoming a potential drug target. Inhibiting HuR could assist in overcoming the major issue of chemoresistance that PDA cancer patients encounter, because to date, no matter of the protocol used, PDA entails one of the most dismal prognoses among cancers.
References
- Maisonneuve, P. Epidemiology and burden of pancreatic cancer. Presse Médicale 2019, 48, e113–e123.
- Sohn, T.A.; Yeo, C.J.; Cameron, J.L.; Koniaris, L.; Kaushal, S.; Abrams, R.A.; Sauter, P.K.; Coleman, J.; Hruban, R.H.; Lillemoe, K.D. Resected adenocarcinoma of the pancreas?616 patients: Results, outcomes, and prognostic indicators. J. Gastrointest. Surg. 2000, 4, 567–579.
- Hahn, S.; Schutte, M.; Kern, S.E.; Hoque, A.T.M.S.; Moskaluk, C.A.; Da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; et al. DPC4, A Candidate Tumor Suppressor Gene at Human Chromosome 18q21.1. Science 1996, 271, 350–353.
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806.
- Seymour, A.B.; Hruban, R.H.; Redston, M.; Caldas, C.; Powell, S.M.; Kinzler, K.W.; Yeo, C.J.; Kern, E.S. Allelotype of pancreatic adenocarcinoma. Cancer Res. 1994, 54, 2761–2764.
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85.
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52.
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A.; The WHO Classification of Tumours Editorial Board. The 2019 WHO classification of tumours of the digestive system. Histopathology 2019, 76, 182–188.
- Conlon, K.C.; Klimstra, D.S.; Brennan, M.F. Long-Term Survival After Curative Resection for Pancreatic Ductal Adenocarcinoma: Clinicopathologic analysis of 5-year survivors. Ann. Surg. 1996, 223, 273–279.
- Neoptolemos, J.P.; Stocken, D.D.; Dunn, J.; Almond, J.; Beger, H.G.; Pederzoli, P.; Bassi, C.; Dervenis, C.; Fernandez-Cruz, L.; Lacaine, F.; et al. Influence of Resection Margins on Survival for Patients with Pancreatic Cancer Treated by Adjuvant Chemoradiation and/or Chemotherapy in the ESPAC-1 Randomized Controlled Trial. Ann. Surg. 2001, 234, 758–768.
- Neoptolemos, J.; Stocken, D.D.; Falconi, M.; Pederzoli, P.; Pap, A.; Spooner, D.; Kerr, D.J.; Büchler, M.W.; Friess, H.; Bassi, C.; et al. A Randomized Trial of Chemoradiotherapy and Chemotherapy after Resection of Pancreatic Cancer. N. Engl. J. Med. 2004, 350, 1200–1210.
- Pandey, V.; Storz, P. Targeting the tumor microenvironment in pancreatic ductal adenocarcinoma. Expert Rev. Anticancer Ther. 2019, 19, 473–482.
- Ren, B.; Cui, M.; Yang, G.; Wang, H.; Feng, M.; You, L.; Zhao, Y. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol. Cancer 2018, 17, 1–15.
- Zhang, Y.-F.; Jiang, S.-H.; Hu, L.-P.; Huang, P.-Q.; Wang, X.; Li, J.; Zhang, X.; Nie, H.-Z.; Zhang, Z.-G. Targeting the tumor microenvironment for pancreatic ductal adenocarcinoma therapy. Chin. Clin. Oncol. 2019, 8, 18.
- Hogan, D.J.; Riordan, D.P.; Gerber, A.P.; Herschlag, D.; Brown, P.O. Diverse RNA-Binding Proteins Interact with Functionally Related Sets of RNAs, Suggesting an Extensive Regulatory System. PLoS Biol. 2008, 6, e255.
- Peng, W.; Furuuchi, N.; Aslanukova, L.; Huang, Y.-H.; Brown, S.Z.; Jiang, W.; Addya, S.; Vishwakarma, V.; Peters, E.; Brody, J.R.; et al. Elevated HuR in Pancreas Promotes a Pancreatitis-Like Inflammatory Microenvironment That Facilitates Tumor Development. Mol. Cell. Biol. 2018, 38, e00427-17.
- Holmes, B.; Benavides-Serrato, A.; Freeman, R.S.; Landon, K.A.; Bashir, T.; Nishimura, R.N.; Gera, J. mTORC2/AKT/HSF1/HuR constitute a feed-forward loop regulating Rictor expression and tumor growth in glioblastoma. Oncogene 2017, 37, 732–743.
- Kurosu, T.; Ohga, N.; Hida, Y.; Maishi, N.; Akiyama, K.; Kakuguchi, W.; Kuroshima, T.; Kondo, M.; Akino, T.; Totsuka, Y.; et al. HuR keeps an angiogenic switch on by stabilising mRNA of VEGF and COX-2 in tumour endothelium. Br. J. Cancer 2011, 104, 819–829.
- Lunde, B.M.; Moore, C.; Varani, G. RNA-binding proteins: Modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490.
- Ma, W.-J.; Chung, S.; Furneaux, H. The Elav-like proteins bind to AU-rich elements and to the poly(A) tail of mRNA. Nucleic Acids Res. 1997, 25, 3564–3569.
- Srikantan, S. HuR function in disease. Front. Biosci. 2012, 17, 189–205.
- Kotta-Loizou, I.; Vasilopoulos, S.N.; Coutts, R.H.; Theocharis, S. Current Evidence and Future Perspectives on HuR and Breast Cancer Development, Prognosis, and Treatment. Neoplasia 2016, 18, 674–688.
- Giaginis, C.; Alexandrou, P.; Tsoukalas, N.; Sfiniadakis, I.; Kavantzas, N.; Agapitos, E.; Patsouris, E.; Theocharis, S. Hu-antigen receptor (HuR) and cyclooxygenase-2 (COX-2) expression in human non-small-cell lung carcinoma: Associations with clinicopathological parameters, tumor proliferative capacity and patients’ survival. Tumor Biol. 2014, 36, 315–327.
- Levidou, G.; Kotta-Loizou, I.; Tasoulas, J.; Papadopoulos, T.; Theocharis, S. Clinical Significance and Biological Role of HuR in Head and Neck Carcinomas. Dis. Markers 2018, 2018, 1–13.
- Kotta-Loizou, I.; Giaginis, C.; Theocharis, S. Clinical significance of HuR expression in human malignancy. Med. Oncol. 2014, 31, 1–19.
- Giaginis, C.; Alexandrou, P.; Delladetsima, I.; Karavokyros, I.; Danas, E.; Giagini, A.; Patsouris, E.; Theocharis, S. Clinical Significance of Hu-Antigen Receptor (HuR) and Cyclooxygenase-2 (COX-2) Expression in Human Malignant and Benign Thyroid Lesions. Pathol. Oncol. Res. 2015, 22, 189–196.
- Goutas, D.; Pergaris, A.; Giaginis, C.; Theocharis, S. HuR as Therapeutic Target in Cancer: What the Future Holds. Curr. Med. Chem. 2021, 28, 1.
- Jimbo, M.; Blanco, F.F.; Huang, Y.-H.; Telonis, A.G.; Screnci, B.A.; Cosma, G.L.; Alexeev, V.; Gonye, G.E.; Yeo, C.J.; Sawicki, J.A.; et al. Targeting the mRNA-binding protein HuR impairs malignant characteristics of pancreatic ductal adenocarcinoma cells. Oncotarget 2015, 6, 27312–27331.
- Costantino, C.L.; Witkiewicz, A.K.; Cozzitorto, J.A.; Kennedy, E.P.; Dasgupta, A.; Keen, J.C.; Yeo, C.J.; Gorospe, M.; Brody, J.R. The Role of HuR in Gemcitabine Efficacy in Pancreatic Cancer: HuR Up-regulates the Expression of the Gemcitabine Metabolizing Enzyme Deoxycytidine Kinase. Cancer Res. 2009, 69, 4567–4572.
- Dixon, D.A.; Tolley, N.D.; King, P.H.; Nabors, L.B.; McIntyre, T.M.; Zimmerman, G.A.; Prescott, S.M. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J. Clin. Investig. 2001, 108, 1657–1665.
- McAllister, F.; Pineda, D.M.; Lankapalli, R.H.; Winter, J.M.; Yeo, C.J.; Witkiewicz, A.K.; A Iacobuzio-Donahue, C.; Laheru, D.; Brody, J.R.; Jimbo, M.; et al. dCK expression correlates with 5-fluorouracil efficacy and HuR cytoplasmic expression in pancreatic cancer: A dual-institutional follow-up with the RTOG 9704 trial. Cancer Biol. Ther. 2014, 15, 688–698.
- Lal, S.; Burkhart, R.A.; Beeharry, N.; Bhattacharjee, V.; Londin, E.; Cozzitorto, J.A.; Romeo, C.; Jimbo, M.; Norris, Z.A.; Yeo, C.J.; et al. HuR Posttranscriptionally Regulates WEE1: Implications for the DNA Damage Response in Pancreatic Cancer Cells. Cancer Res. 2014, 74, 1128–1140.
- Lal, P.; Cerofolini, L.; D’Agostino, V.G.; Zucal, C.; Fuccio, C.; Bonomo, I.; Dassi, E.; Giuntini, S.; Di Maio, D.; Vishwakarma, V.; et al. Regulation of HuR structure and function by dihydrotanshinone-I. Nucleic Acids Res. 2017, 45, 9514–9527.
- Fan, J.; Yang, X.; Wang, W.; Wood, W.H.; Becker, K.; Gorospe, M. Global analysis of stress-regulated mRNA turnover by using cDNA arrays. Proc. Natl. Acad. Sci. USA 2002, 99, 10611–10616.
- Cheadle, C.; Fan, J.; Cho-Chung, Y.S.; Werner, T.; Ray, J.; Do, L.; Gorospe, M.; Becker, K.G. Control of gene expression during T cell activation: Alternate regulation of mRNA transcription and mRNA stability. BMC Genom. 2005, 6, 75.
- Koong, A.; Mehta, V.K.; Le, Q.T.; Fisher, G.A.; Terris, D.J.; Brown, J.; Bastidas, A.J.; Vierra, M. Pancreatic tumors show high levels of hypoxia. Int. J. Radiat. Oncol. 2000, 48, 919–922.
- Vineis, P. Exposures, mutations and the history of causality. J. Epidemiol. Community Health 2000, 54, 652–653.
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703.
- Burkhart, A.R.; Pineda, D.M.; Chand, S.N.; Romeo, C.; Londin, E.R.; Karoly, E.D.; Cozzitorto, A.J.; Rigoutsos, I.; Yeo, C.J.; Brody, J.R.; et al. HuR is a post-transcriptional regulator of core metabolic enzymes in pancreatic cancer. RNA Biol. 2013, 10, 1312–1323.
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975.
- Blanco, F.F.; Jimbo, M.; Wulfkuhle, J.D.; Gallagher, I.J.; Deng, J.; Enyenihi, L.; Meisner-Kober, N.; Londin, E.; Rigoutsos, I.; Sawicki, A.J.; et al. The mRNA-binding protein HuR promotes hypoxia-induced chemoresistance through posttranscriptional regulation of the proto-oncogene PIM1 in pancreatic cancer cells. Oncogene 2016, 35, 2529–2541.
- Zarei, M.; Lal, S.; Parker, S.; Nevler, A.; Vaziri-Gohar, A.; Dukleska, K.; Mambelli-Lisboa, N.C.; Moffat, C.; Blanco, F.F.; Chand, S.N.; et al. Posttranscriptional Upregulation of IDH1 by HuR Establishes a Powerful Survival Phenotype in Pancreatic Cancer Cells. Cancer Res. 2017, 77, 4460–4471.
- Chang, Q.; Jurisica, I.; Do, T.; Hedley, D. Hypoxia Predicts Aggressive Growth and Spontaneous Metastasis Formation from Orthotopically Grown Primary Xenografts of Human Pancreatic Cancer. Cancer Res. 2011, 71, 3110–3120.
- Büchler, P.; Reber, H.A.; Büchler, M.W.; Friess, H.; Lavey, R.S.; Hines, O.J. Antiangiogenic activity of genistein in pancreatic carcinoma cells is mediated by the inhibition of hypoxia-inducible factor-1 and the down-regulation ofVEGFgene expression. Cancer 2003, 100, 201–210.
- Costello, E. The role of inflammatory cells in fostering pancreatic cancer cell growth and invasion. Front. Physiol. 2012, 3, 270.
- Yako, Y.Y.; Kruger, D.; Smith, M.; Brand, M. Cytokines as Biomarkers of Pancreatic Ductal Adenocarcinoma: A Systematic Review. PLoS ONE 2016, 11, e0154016.
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nat. Cell Biol. 2017, 542, 177–185.
- Porta, C.; Marino, A.; Consonni, F.M.; Bleve, A.; Mola, S.; Storto, M.; Riboldi, E.; Sica, A. Metabolic influence on the differentiation of suppressive myeloid cells in cancer. Carcinogenesis 2018, 39, 1095–1104.
- Cani, P.D.; Jordan, B.F. Gut microbiota-mediated inflammation in obesity: A link with gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 671–682.
- Zitvogel, L.; Pietrocola, F.; Kroemer, G. Nutrition, inflammation and cancer. Nat. Immunol. 2017, 18, 843–850.
- Yiakouvaki, A.; Dimitriou, M.; Karakasiliotis, I.; Eftychi, C.; Theocharis, S.; Kontoyiannis, D.L. Myeloid cell expression of the RNA-binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis. J. Clin. Investig. 2012, 122, 48–61.
- Chae, M.-J.; Sung, H.Y.; Kim, E.-H.; Lee, M.; Kwak, H.; Chae, C.H.; Kim, S.; Park, W.-Y. Chemical inhibitors destabilize HuR binding to the AU-rich element of TNF-α mRNA. Exp. Mol. Med. 2009, 41, 824–831.
- Dhir, T.; Schultz, C.W.; Jain, A.; Brown, S.Z.; Haber, A.; Goetz, A.; Xi, C.; Su, G.; Xu, L.; Posey, J.; et al. Abemaciclib Is Effective Against Pancreatic Cancer Cells and Synergizes with HuR and YAP1 Inhibition. Mol. Cancer Res. 2019, 17, 2029–2041.
- Zhou, H.; Telonis, A.G.; Jing, Y.; Xia, N.L.; Biederman, L.; Jimbo, M.; Blanco, F.; Londin, E.; Brody, J.R.; Rigoutsos, I. GPRC5A is a potential oncogene in pancreatic ductal adenocarcinoma cells that is upregulated by gemcitabine with help from HuR. Cell Death Dis. 2016, 7, e2294.
- Romeo, C.; Weber, M.C.; Zarei, M.; Decicco, D.; Chand, S.N.; Lobo, A.D.; Winter, J.M.; Sawicki, J.A.; Sachs, J.N.; Meisner-Kober, N.; et al. HuR Contributes to TRAIL Resistance by Restricting Death Receptor 4 Expression in Pancreatic Cancer Cells. Mol. Cancer Res. 2016, 14, 599–611.
- Jain, A.; Agostini, L.C.; Mccarthy, G.; Chand, S.N.; Ramirez, A.; Nevler, A.; Cozzitorto, J.; Schultz, C.W.; Lowder, C.Y.; Smith, K.M.; et al. Poly (ADP) Ribose Glycohydrolase Can Be Effectively Targeted in Pancreatic Cancer. Cancer Res. 2019, 79, 4491–4502.
- Chand, S.N.; Zarei, M.; Schiewer, M.J.; Kamath, A.R.; Romeo, C.; Lal, S.; Cozzitorto, J.A.; Nevler, A.; Scolaro, L.; Londin, E.; et al. Posttranscriptional Regulation of PARG mRNA by HuR Facilitates DNA Repair and Resistance to PARP Inhibitors. Cancer Res. 2017, 77, 5011–5025.
- Liu, J.F.; Xiong, N.; Campos, S.M.; Wright, A.A.; Krasner, C.; Schumer, S.; Horowitz, N.; Veneris, J.; Tayob, N.; Morrissey, S.; et al. Phase II Study of the WEE1 Inhibitor Adavosertib in Recurrent Uterine Serous Carcinoma. J. Clin. Oncol. 2021, 39, 1531–1539.
- Cole, K.A.; Pal, S.; Kudgus, R.A.; Ijaz, H.; Liu, X.; Minard, C.G.; Pawel, B.R.; Maris, J.M.; Haas-Kogan, D.A.; Voss, S.D.; et al. Phase I Clinical Trial of the Wee1 Inhibitor Adavosertib (AZD1775) with Irinotecan in Children with Relapsed Solid Tumors: A COG Phase I Consortium Report (ADVL1312). Clin. Cancer Res. 2020, 26, 1213–1219.
- Kong, A.; Good, J.; Kirkham, A.; Savage, J.; Mant, R.; Llewellyn, L.; Parish, J.; Spruce, R.; Forster, M.; Schipani, S.; et al. Phase I trial of WEE1 inhibition with chemotherapy and radiotherapy as adjuvant treatment, and a window of opportunity trial with cisplatin in patients with head and neck cancer: The WISTERIA trial protocol. BMJ Open 2020, 10, e033009.
- Leijen, S.; Van Geel, R.M.J.M.; Sonke, G.; De Jong, D.; Rosenberg, E.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361.
- Cuneo, K.C.; Morgan, M.A.; Sahai, V.; Schipper, M.J.; Parsels, L.A.; Parsels, J.D.; Devasia, T.; Al-Hawaray, M.; Cho, C.S.; Nathan, H.; et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination With Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. J. Clin. Oncol. 2019, 37, 2643–2650.
- Leijen, S.; Van Geel, R.M.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.A.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4371–4380.
- Kausar, T.; Schreiber, J.S.; Karnak, D.; Parsels, L.A.; Parsels, J.D.; Davis, M.A.; Zhao, L.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Sensitization of Pancreatic Cancers to Gemcitabine Chemoradiation by WEE1 Kinase Inhibition Depends on Homologous Recombination Repair. Neoplasia 2015, 17, 757–766.
- Hartman, S.J.; Bagby, S.M.; Yacob, B.W.; Simmons, D.M.; MacBeth, M.; Lieu, C.H.; Davis, S.L.; Leal, A.D.; Tentler, J.J.; Diamond, J.R.; et al. WEE1 Inhibition in Combination With Targeted Agents and Standard Chemotherapy in Preclinical Models of Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2021, 11, 957.
- Zhou, H.; Rigoutsos, I. The emerging roles of GPRC5A in diseases. Oncoscience 2014, 1, 765–776.
- Johnstone, R.W.; Frew, A.J.; Smyth, M. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer 2008, 8, 782–798.
- Pineda, D.M.; Rittenhouse, D.W.; Valley, C.C.; Cozzitorto, J.A.; Burkhart, R.A.; Leiby, B.; Winter, J.M.; Weber, M.C.; Londin, E.R.; Rigoutsos, I.; et al. HuR’s post-transcriptional regulation of death receptor 5 in pancreatic cancer cells. Cancer Biol. Ther. 2012, 13, 946–955.
- Lemke, J.; Noack, A.; Adam, D.; Tchikov, V.; Bertsch, U.; Röder, C.; Schütze, S.; Wajant, H.; Kalthoff, H.; Trauzold, A. TRAIL signaling is mediated by DR4 in pancreatic tumor cells despite the expression of functional DR5. J. Mol. Med. 2010, 88, 729–740.
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of Poly (ADP-ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28.
- Bouwman, P.; Jonkers, J. Molecular Pathways: How Can BRCA-Mutated Tumors Become Resistant to PARP Inhibitors? Clin. Cancer Res. 2014, 20, 540–547.
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.-C.; Choi, Y.-E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046.
- Tatarian, T.; Jiang, W.; Halloran, C.; Palmer, D.; Buchler, M.; Yeo, C.J.; Winter, J.M.; Brody, J.R.; Leiby, B.E.; Grigoli, A.; et al. Cytoplasmic HuR Status Predicts Disease-free Survival in Resected Pancreatic Cancer: A Post-hoc Analysis From the International Phase III ESPAC-3 Clinical Trial. Ann. Surg. 2018, 267, 364–369.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
558
Revisions:
2 times
(View History)
Update Date:
24 Sep 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No