Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pedro Bullon | + 2355 word(s) | 2355 | 2021-09-23 10:06:03 | | | |
| 2 | Vivi Li | Meta information modification | 2355 | 2021-09-24 03:18:02 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bullon, P. Diabetes Mellitus and Periodontitis. Encyclopedia. Available online: https://encyclopedia.pub/entry/14479 (accessed on 28 July 2026).
Bullon P. Diabetes Mellitus and Periodontitis. Encyclopedia. Available at: https://encyclopedia.pub/entry/14479. Accessed July 28, 2026.
Bullon, Pedro. "Diabetes Mellitus and Periodontitis" Encyclopedia, https://encyclopedia.pub/entry/14479 (accessed July 28, 2026).
Bullon, P. (2021, September 23). Diabetes Mellitus and Periodontitis. In Encyclopedia. https://encyclopedia.pub/entry/14479
Bullon, Pedro. "Diabetes Mellitus and Periodontitis." Encyclopedia. Web. 23 September, 2021.
Copy Citation
Diabetes and periodontitis are two of the most prevalent diseases worldwide that negatively impact the quality of life of the individual suffering from them. They are part of the chronic inflammatory disease group or, as recently mentioned, non-communicable diseases, with inflammation being the meeting point among them. Inflammation hitherto includes vascular and tissue changes, but new technologies provide data at the intracellular level that could explain how the cells respond to the aggression more clearly.
periodontal disease
diabetes
mitochondrial dysfunction
oxidative stress
AMPK
autophagy
1. Introduction
Diabetes is a chronic metabolic disease characterized by hyperglycemia and is one of the leading causes of death worldwide among non-communicable diseases [1]. Periodontitis is the advanced form of periodontal disease and is one of the most prevalent diseases in the world. It is mostly caused by oral microbiota dysbiosis [2][3], but some risk factors also impact its development such as diabetes, smoking and genetic predisposition [4]. Diabetes is an important grade modifier used as an indicator of the rate of periodontitis progression [5].
Inflammation marks the link between diabetes and periodontal disease [6]. However, some authors consider that there is still scarce information based on research with representative samples and prospective longitudinal studies [7]. It is important to note that it is difficult to perform a longitudinal study and to define whether the pathological associations are causal in nature in chronic inflammatory diseases studies.
Hitherto inflammation is described as vascular and tissue alterations. An aggression produces some cytokines that induces the extravasation of plasma and blood cells that try to control and restore the damage. Periodontitis and T2DM, independently, have elevated inflammatory markers. However, when present at the same time, there is an exacerbation of this immunoinflammatory response. M1-type macrophage [8][9][10], neutrophils [11][12][13][14], and polymorphonuclear cells (PMN) [15] usually have their function upregulated, and dendritic cells are reduced or immature [16]. Consequently, pro-inflammatory cytokines such as IL-1β, IL-17, IL-6, TNFα, INFγ used to be higher and IL-10 reduced [17][18][19][20][21]. This scenario favors the great tissue destruction observed in periodontal tissues, micro and macrovascular lesions, lipid profile alterations (high low-density lipoprotein (LDL) and triglycerides) and difficulty in glycemic control [22][23][24][25].
All these aforementioned mechanisms explain the pathogenesis at the tissue level. Tissues are made up of cells that mediate the immunity and suffer the aggression. New technological advances allow us to study deeply the cell mechanisms involved. Therefore, it is essential to understand not only the pathological alterations at the tissue level, but also the intracellular molecular mechanisms involved in this process that usually occur under subclinical conditions even before a state of complete inflammatory disease is established. Preclinical studies have been helpful in understanding the basic mechanisms involved in the onset of diabetes, periodontitis, and their systemic effects [26].
2. Biological Membrane Alteration
Plasma membrane is a highly dynamic structure composed of phospholipid bilayer and lipid rafts, being one of the main structures of all living systems, delimiting cells and organelles such as lysosomes and mitochondria. Lipid rafts are composed of cholesterol, glycosphingolipids, and specific proteins, which are associated and dissociated in the second scale [27][28][29].
These microdomains are involved in cellular signaling and membrane permeability, such as endo- and exocytosis during bacterial or toxin aggression [30][31], immune cell activation [32], redox signaling [33][34], osteoclastogenesis induction [28][35], and insulin secretion and sensitivity [32][36]. The type and amount of lipids vary in each cell membrane according to their function and the individual’s diet and are influenced by lipid metabolism in health and disease condition. Disruption of this structure may alter several physiological cellular functions [27][28][29].
Fatty acids (FA) are important membrane structural components and signaling molecules, and any change in their length or degree of saturation can directly impact membrane plasticity. High concentration of saturated FA (SFA) induces negative effects on the plasma membrane by increasing its fluidity and activity, increasing toll-like receptor (TLR) signaling translocation and RANKL activation [28][37][38].
Patients with diabetes have high levels of SFA and overexpression of TLR4/CD36-mediated pathway in gingival fibroblasts [30][39]. Palmitate (saturated) is enhanced in hyperglycemia, and it is even higher in the presence of P. gingivalis [39], which is in agreement with the increase of FA uptake by lipid rafts after periodontal lipopolysaccharides (LPS) stimulation [40], suggesting an exacerbation of inflammation in individuals with T2DM and periodontitis.
Polyunsaturated FA (PUFA) play a role in modulating mitochondrial function, inflammatory response, improving hormone sensitivity, especially insulin, and enhancing membrane fluidity and responsiveness. PUFA may present a pro-inflammatory profile (in case omega-6 prevail) in the initial phases of inflammatory response or an anti-inflammatory profile (with omega-3 being the most represented) during the resolution of inflammation. Omega-3 seems to inhibit factor nuclear kappa B (NF-κβ) activation and TLR dimerization, which reduces SFA pro-inflammatory stimulus [29][37][39][41][42], and it has been also related to clinical and immunological benefits for patients with T2DM after daily supplementation and periodontal debridement [43].
Integrity of membrane properties have been associated with a diet rich in unsaturated fats such as olive oil [44], while high-fat diet (rich in saturated fat and cholesterol) seems to alter cellular properties and exacerbate the inflammatory response and increase hyperlipidemia and alveolar bone loss in periodontitis models [45]. The inhibition of specific glycosphingolipids of lipid rafts improved glucose control, insulin sensitivity in T2DM patients [36], and prevent RANKL-osteoclast induction [33][35].
Lipid peroxidation (LPO) is an oxidative degradation of membrane lipids, which is increased in diabetes owing to the alteration of oxidative metabolism and the overproduction of reactive oxygen species (ROS) [46]. This reaction produces lipid peroxides that bind to proteins and create unstable lipid radicals. Repeated cycles of LPO can activate the NF-κβ pathway, inducing a pro-inflammatory response, contributing to maintaining oxidative stress and causing serious damage to cell membranes [46].
The intensity of LPO strongly depends on the degree of lipid unsaturation and this reaction is amplified as long as oxygen and unoxidized PUFA are available [47]. Lipid marker alterations have been associated with the severity of periodontitis and uncontrolled T2DM. Several LPO markers are used to monitor ROS production, and they are positively associated with cytokines’ local and systemic expression in patients with T2DM and periodontitis with dyslipidemia, which is even worse in poorly controlled T2DM [46] (Figure 1).
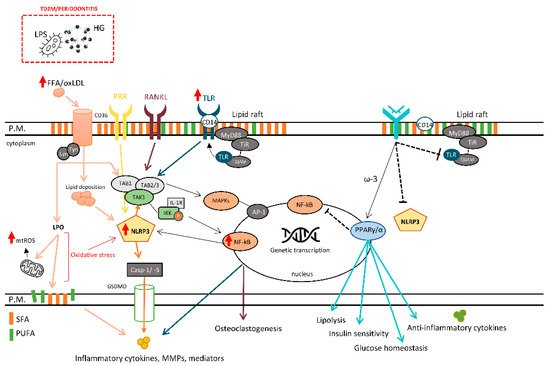
Figure 1. The role of plasma membrane and its influence on different cellular responses. LPS, lipopolysaccharides; FFA, free fatty acid; oxLDL, oxidized LDL; LPO, lipid peroxidation; mtROS, mitochondrial reactive oxygen species; CD14/36, cluster of differentiation 14/36; PRR, pattern recognition receptors; RANKL, receptor activator of nuclear factor-kappa beta ligand; TLR, toll-like receptor; IKK, inhibitor of kappa B kinase; IL-1R, interleukin-1 receptor; MyD88, myeloid differentiation primary response 88; NF-κB, nuclear factor kappa B; TAB1/2/3, transforming growth factor beta (TGF-β) activated kinase 1-binding protein 1/2/3; TAK1, TGF-β activated kinase 1; NLRP3, NLR family pyrin domain-containing protein 3 inflammasome; Casp-1/-5, caspase-1/5; MAPK, mitogen-activated protein kinases; AP-1, Activator protein 1; MMPs, matrix metalloproteinases; ω-3, ômega-3 polyunsaturated fatty acid; PUFA, polyunsaturated fatty acid; SFA, saturated fatty acid; HG, high glucose.
Currently, T2DM patients have dyslipidemia, an imbalance of body lipids characterized by high levels of triglycerides and LDL and low levels of high-density lipoprotein (HDL). This condition increases oxidative metabolism and LPO, thereby maintaining a vicious cycle of chronic pro-inflammatory condition [46]. The disturbance of glycemic metabolism and the continued activation of the polyol pathway to metabolize the excess of glucose also causes membrane alterations, increases LPO, and coupled with reduced antioxidant system (AOX) [48] upregulates immune cell responses and visceral adiposity [49]. Transcriptome analysis has facilitated the evidence of deregulation of different inflammatory molecular pathways, by co-expressed genes, in association with the quality of adipose tissue and type 2 diabetes [50].
3. Aggression Recognition
Humans are multicellular organisms, and it is essential to distinguish between our own cells and others that can be harmful, as well as physical and chemical factors, through membrane cell receptors that stimulate immunity. These are fundamental sensory elements for host defense that can be stimulated by hormones and inflammatory mediators.
The immune system recognizes aggression through the connection of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) to pattern recognition receptors (PRRs). These PRRs can be TLRs, NOD-like receptors (NLRs), RAGE, C-type lectin receptors (CLRs), and complement receptors. The increased levels of interleukin (IL)-1β enhance the expression of some cell receptors such as TLR4 which are involved in the signaling and activation of NF-κβ and mitogen-activated protein kinase (MAPK) pathways [51].
Inflammasomes, the key regulators of innate, adaptive, and host responses, are a cytoplasmic multi-protein complex composed of NLRs and different types of proteins. NIMA-related kinase 7 (Nek7) is an indispensable upstream factor involved in NLR family pyrin domain-containing protein 3 (NLRP3) inflammasome formation and regulates the release of pro-inflammatory cytokines. The inflammasome complex activates caspase-1 and -5, which consequently release the first cytokines IL-1β and IL-18 against PAMP or DAMP, producing a cascade of local and systemic responses [52].
Inflammasomes are activated and modulated by different metabolic alterations, and it has been reported that P. gingivalis infection induces an overexpression of PRRs and NLRP3 in T2DM-periodontitis patients, along with caspase-1 and IL-1β [51][53]. The most recently discovered innate immune cells on the periodontal tissue of periodontitis patients and mice-models of periodontitis are the innate lymphoid cells (ILCs). They are activated by PAMPs and DAMPs and play a role on initiation, modulation, and resolution of inflammation through cytokine release [54]. Adenosine monophosphate-activated protein kinase (AMPK) acts as a modulator of ILCs function [54] and NLRP3 [55], reducing their negative effects.
Hyperglycemia, even in intermittent periods, exacerbates TLR4 and RAGE expression [56]. The disease severity has been related to elevations in pro-inflammatory cytokine expression and their involvement in the increased expression of RAGE or TLR4 on the surface of epithelial cells, fibroblasts, and macrophages [56]. A significant association between RAGE polymorphism and patients with periodontitis and T2DM exists, but no association was observed in patients with only periodontitis [57]. However, it is difficult to establish whether this polymorphism can be considered a risk factor related to the development of periodontitis when associated with T2DM or if this genetic alteration is just linked to diabetes. Further investigations in patients with diabetes but without periodontitis are necessary to confirm this risk [26].
Recently, polymorphisms of TNF-α, TNFR1, TNFR2 and lymphotoxin-α were evaluated: no SNP was found to be a cross-susceptibility factor between periodontitis and T2DM. Therefore, the development of periodontitis in T2DM may be related to pathological alterations in the periodontium caused by diabetes due to hyperglycemia, high AGE levels and oxidative stress. T2DM is suggested to mask the impact of periodontitis on systemic inflammation [58].
The entire transcriptional profile of LPS of P. gingivalis in PDL cells has been recently described, and 36 differentially expressed genes (DEGs) have been identified in PDLs cultured with LPS for 24 h and 72 h. It was possible to observe that different biological processes, molecular functions, and cellular components are involved in the initiation and progression of periodontitis [59]. Additionally, dysregulation of immunoactivation mechanisms of neutrophils and B cells were evidenced by differentially expressed genes [60][61].
4. Mitochondrial Dysfunction
Oxidative phosphorylation by mitochondria is responsible for most of the ATP produced and ROS production also appears [62]. ROS release at early stages is adaptative, acting as important signaling molecules after an aggression, and is controlled by intracellular redox status through AOX [52]. However, at high concentrations they cause cellular lesions [50].
The excess of electron donors in the mitochondrial electron transport chain is one of the main factors responsible for NADH/NAD+ redox imbalance, because as more electrons are transported, the higher the ROS production [63]. In metabolic disorders, positive feedback is established with the increased release of ROS which stimulates the neighboring mitochondria to control the excess of these molecules, resulting in more ROS production [25][62].
Mitochondrial dysfunction is considered the major source of ROS causing damage to all cellular components and disrupting the normal signaling mechanisms. Altogether, these effects directly impaired the inflammatory response, inducing a pro-inflammatory state [25][62]. Oxidative stress arises and is maintained due to the increase in mitochondrial ROS production and inefficient (or absence) of enough AOX levels, resulting in an imbalance of the cellular redox state [64][65].
Advanced glycation end products (AGEs) arise from non-enzymatic glycation and oxidation of proteins and lipids [66]. They cause cellular damage by modifying protein function and cellular interaction with the extracellular membrane, alter the intracellular Ca2+ concentration and mitochondrial function, deregulate the inflammatory response, influence wound repair, and increase oxidative stress through the connection with its receptors, RAGE [6][66]. It has been suggested that high levels of AGEs may modify collagen structure, making the periodontal tissues less soluble with less reparative tendency, and along with other altered cellular responses, making them more susceptible to periodontal breakdown [67].
The degradation of AGEs occurs intracellularly by endocytosis and lysosomal activity, and galectin-3 have been discovered to be an essential molecule to AGEs removal [68][69]. In addition, low levels of galectin-3 have been associated with deficiency in glucose uptake, endothelial dysfunction in a diabetic mice model [70], and increased bone loss under high glucose condition and periodontal/LPS infection [69], which are negatively regulated by micro-RNA-124-3p [69]. Patients with diabetes used to have high levels of AGE and RAGE in human gingival fibroblast which may explain the accelerated periodontitis observed in these patients in accordance with the previous studies [71][72].
Oxidative stress and the AGE-RAGE connection stimulate signaling pathways, such as MAPK and NF-κβ, with subsequent pro-inflammatory gene transcription and increased ROS production in endothelial cells, vascular smooth muscle cells, and macrophages. The high number and activity of immune cells, mostly by the excessive response of phagocytes during hyperinflammatory response, contribute to the overall cellular stress [11][65][73]. Patients with T2DM and periodontitis showed higher levels of AGEs and ROS production than healthy individuals [11]. This oxidative stress is induced even when both diseases are not present simultaneously; however, when they are together, it becomes more severe [24].
Mitochondrial dysfunction and high mitochondrial ROS production [25][62] result in cellular stress at the molecular level, causing a reduction in protein expression, loss of mitochondrial mass, and impaired membrane potential [62], and these alterations are present in diabetes and periodontitis. Moreover, the accumulation of mitochondrial DNA (mtDNA) alterations such as mtDNA heteroplasmy and copy number, noncoding ribonucleic acid (RNA), epigenetic modification of the mitochondrial genome, epitranscriptomic regulation of the mtDNA-encoded mitochondrial transcriptome and mtDNA mutations and polymorphisms have been related to endothelial dysfunction, change in metabolism of the liver, adipose tissue, myocardium, and skeletal muscles, and poor metabolic control [74][75][76][77][78]. These parameters could be used as markers to characterize the dysregulated immune-inflammatory response commonly detected in individuals with periodontitis and T2DM [25][76].
References
- International Diabetes Federation. IDF Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019.
- Socransky, S.S.; Haffajee, A.D. Dental biofilms: Difficult therapeutic targets. Periodontology 2002, 28, 12–55.
- Ganesan, S.M.; Joshi, V.; Fellows, M.; Dabdoub, S.M.; Nagaraja, H.N.; O’Donnell, B.; Deshpande, N.R.; Kumar, P.S. A tale of two risks: Smoking, diabetes and the subgingival microbiome. ISME J. 2017, 11, 2075–2089.
- Lang, N.P.; Bartold, P.M. Periodontal health. J. Periodontol. 2018, 89, S9–S16.
- Papapanou, P.N.; Sanz, M.; Buduneli, N.; Dietrich, T.; Feres, M.; Fine, D.H.; Flemmig, T.F.; Garcia, R.; Giannobile, W.V.; Graziani, F.; et al. Periodontitis: Consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J. Periodontol. 2018, 89, S173–S182.
- Verhulst, M.J.L.; Loos, B.G.; Gerdes, V.E.A.; Teeuw, W.J. Evaluating all potential oral complications of diabetes mellitus. Front. Endocrinol. (Lausanne) 2019, 10, 56.
- Nascimento, G.G.; Leite, F.R.M.; Vestergaard, P.; Scheutz, F.; Lopez, R.; López, R. Does diabetes increase the risk of periodontitis? A systematic review and meta-regression analysis of longitudinal prospective studies. Acta Diabetol. 2018, 55, 653–667.
- Almubarak, A.; Tanagala, K.K.K.; Papapanou, P.N.; Lalla, E.; Momen-Heravi, F. Disruption of Monocyte and Macrophage Homeostasis in Periodontitis. Front. Immunol. 2020, 11, 1–11.
- Wang, Q.; Nie, L.; Zhao, P.; Zhou, X.; Ding, Y.; Chen, Q.; Wang, Q. Diabetes fuels periodontal lesions via GLUT1-driven macrophage inflammaging. Int. J. Oral Sci. 2021, 13, 1–20.
- Xu, R.; Zeng, G.; Wang, S.; Tao, H.; Ren, L.; Zhang, Z.; Zhang, Q.; Zhao, J.; Gao, J.; Li, D. Periodontitis promotes the diabetic development of obese rat via miR-147 induced classical macrophage activation. Biomed. Pharmacother. 2016, 83, 892–897.
- Karima, M.; Kantarci, A.; Ohira, T.; Hasturk, H.; Jones, V.L.; Nam, B.-H.; Malabanan, A.; Trackman, P.C.; Badwey, J.A.; van Dyke, T.E. Enhanced superoxide release and elevated protein kinase C activity in neutrophils from diabetic patients: Association with periodontitis. J. Leukoc. Biol. 2005, 78, 862–870.
- Joshi, M.B.; Ahamed, R.; Hegde, M.; Nair, A.S.; Ramachandra, L.; Satyamoorthy, K. Glucose induces metabolic reprogramming in neutrophils during type 2 diabetes to form constitutive extracellular traps and decreased responsiveness to lipopolysaccharides. BBA—Mol. Basis Dis. J. 2020, 1866, 165940.
- Vitkov, L.; Munoz, L.E.; Knopf, J.; Schauer, C.; Oberthaler, H.; Minnich, B.; Hannig, M.; Herrmann, M. Connection between periodontitis-induced low-grade endotoxemia and systemic diseases: Neutrophils as protagonists and targets. Int. J. Mol. Sci. 2021, 22, 4647.
- Herrmann, J.M.; Sonnenschein, S.K.; Groeger, S.E.; Ewald, N.; Arneth, B.; Meyle, J. Refractory neutrophil activation in type 2 diabetics with chronic periodontitis. J. Periodontal Res. 2020, 55, 315–323.
- Manosudprasit, A.; Kantarci, A.; Hasturk, H.; Stephens, D.; Van Dyke, T.E. Spontaneous PMN apoptosis in type 2 diabetes and the impact of periodontitis. J. Leukoc. Biol. 2017, 102, 1431–1440.
- De Rabelo, M.S.; El-Awady, A.; Foz, A.M.; Gomes, G.H.; Rajendran, M.; Meghil, M.M.; Lowry, S.; Romito, G.A.; Cutler, C.W.; Susin, C. Influence of T2DM and prediabetes on blood DC subsets and function in subjects with periodontitis. Oral Dis. 2019, 25, 2020–2029.
- Grauballe, M.B.; Østergaard, J.A.; Schou, S.; Flyvbjerg, A.; Holmstrup, P. Effects of TNF-α blocking on experimental periodontitis and type 2 diabetes in obese diabetic Zucker rats. J. Clin. Periodontol. 2015, 42, 807–816.
- Yoon, A.J.; Cheng, B.; Philipone, E.; Turner, R.; Lamster, I.B. Inflammatory biomarkers in saliva: Assessing the strength of association of diabetes mellitus and periodontal status with the oral inflammatory burden. J. Clin. Periodontol. 2012, 39, 434–440.
- Miranda, T.S.; Almeida, M.L.; Marins, L.M.; da Silva, H.D.P.; Feres, M.; Duarte, P.M. Might smoking assuage the pro-inflammatory effect of diabetes in periodontal sites? Oral Dis. 2020, 26, 200–212.
- Duarte, P.M.; Bezerra, J.P.; Miranda, T.S.; Feres, M.; Chambrone, L.; Shaddox, L.M. Local levels of inflammatory mediators in uncontrolled type 2 diabetic subjects with chronic periodontitis. J. Clin. Periodontol. 2014, 41, 11–18.
- Elazazy, O.; Amr, K.; Abd, A.; Fattah, E.; Abouzaid, M. Evaluation of serum and gingival crevicular fluid microRNA-223, microRNA-203 and microRNA-200b expression in chronic periodontitis patients with and without diabetes type 2. Arch. Oral Biol. 2021, 121, 1–7.
- Ramirez-Tortosa, M.C.; Quiles, J.L.; Battino, M.; Granados, S.; Morillo, J.M.; Bompadre, S.; Newman, H.N.; Bullon, P. Periodontitis is associated with altered plasma fatty acids and cardiovascular risk markers. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 133–139.
- Xu, T.; Brandmaier, S.; Messias, A.C.; Herder, C.; Draisma, H.H.M.; Demirkan, A.; Yu, Z.; Ried, J.S.; Haller, T.; Heier, M.; et al. Effects of metformin on metabolite profiles and LDL cholesterol in patients with type 2 diabetes. Diabetes Care 2015, 38, 1858–1867.
- Sugiyama, S.; Takahashi, S.S.; Tokutomi, F.A.; Yoshida, A.; Kobayashi, K.; Yoshino, F.; Wada-Takahashi, S.; Toyama, T.; Watanabe, K.; Hamada, N.; et al. Gingival vascular functions are altered in type 2 diabetes mellitus model and/or periodontitis model. J. Clin. Biochem. Nutr. 2012, 51, 108–113.
- Masi, S.; Orlandi, M.; Parkar, M.; Bhowruth, D.; Kingston, I.; O’Rourke, C.; Virdis, A.; Hingorani, A.; Hurel, S.J.; Donos, N.; et al. Mitochondrial oxidative stress, endothelial function and metabolic control in patients with type II diabetes and periodontitis: A randomised controlled clinical trial. Int. J. Cardiol. 2018, 271, 263–268.
- Grauballe, M.B.; Østergaard, J.A.; Schou, S.; Flyvbjerg, A.; Holmstrup, P. Blockade of RAGE in Zucker obese rats with experimental periodontitis. J. Periodontal Res. 2017, 52, 97–106.
- Nordzieke, D.E.; Medraño-Fernandez, I. The Plasma Membrane: A Platform for Intra- and Intercellular Redox Signaling. Antioxidants 2018, 7, 168.
- Ersek, A.; Karadimitris, A.; Horwood, N.J. Effect of glycosphingolipids on osteoclastogenesis and osteolytic bone diseases. Front. Endocrinol. (Lausanne) 2012, 3, 1–7.
- Robinson, G.A.; Waddington, K.E.; Pineda-Torra, I.; Elizabeth, C.J. Transcriptional Regulation of T-Cell Lipid Metabolism: Implications for Plasma Membrane Lipid Rafts and T-Cell Function. Front. Immunol. 2017, 8, 1–10.
- Bullón, P. New Theories and Their Clinical Relevance to the Onset and Development of Periodontal Diseases. In Studies on Periodontal Disease; Ekuni, D., Battino, M., Tomofuji, T., Putnins, E.E., Eds.; Springer: New York, NY, USA, 2014; pp. 227–248.
- Lally, E.T.; Boesze-Battaglia, K.; Dhingra, A.; Gomez, N.M.; Lora, J.; Mitchell, C.H.; Giannakakis, A.; Fahim, S.A.; Benz, R.; Balashova, N. Aggregatibacter actinomycetemcomitans LtxA Hijacks Endocytic Trafficking Pathways in Human Lymphocytes. Pathogens 2020, 9, 74.
- Chentouf, M.; Guzman, C.; Hamze, M.; Gross, R.; Lajoix, A.D.; Peraldi-Roux, S. Possible protective effect of membrane lipid rafts against interleukin-1β-mediated anti-proliferative effect in INS-1 cells. PLoS ONE 2014, 9, e102889.
- Dumitru, C.A.; Zhang, Y.; Li, X.; Gulbins, E. Ceramide: A Novel Player in Reactive Oxygen Species-Induced Signaling? Antioxid. Redox Signal. 2007, 9, 1535–1540.
- Zang, A.Y.; Yi, F.; Zhang, G.; Gulbins, E.; Li, P.L. Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension 2006, 47, 74–80.
- Liao, H.-J.; Tsai, H.-F.; Wu, C.-S.; Chyuan, L.-T.; Hsu, P.-N. TRAIL inhibits RANK signaling and suppresses osteoclast activation via inhibiting lipid raft assembly and TRAF6 recruitment. Cell Death Dis. 2019, 10, 1–11.
- Zhao, H.; Przybylska, M.; Wu, I.-H.; Zhang, J.; Siegel, C.; Komarnitsky, S.; Yew, N.S.; Cheng, S.H. Inhibiting glycosphingolipid synthesis improves glycemic control and insulin sensitivity in animal models of type 2 diabetes. Diabetes 2007, 56, 1210–1218.
- Hwang, D.H.; Kim, J.-A.; Lee, J.Y.; Hwang, D.H. Mechanisms for the activation of Toll-like receptor 2/4 by saturated fatty acids and inhibition by docosahexaenoic acid HHS Public Access. Eur. J. Pharmacol. 2016, 785, 24–35.
- Li, Y.; Guan, J.; Wang, W.; Hou, C.; Zhou, L.; Ma, J.; Cheng, Y.; Jiao, S.; Zhou, Z. TRAF3-interacting JNK-activating modulator promotes inflammation by stimulating translocation of Toll-like receptor 4 to lipid rafts. J. Biol. Chem. 2019, 294, 2744–2756.
- Shikama, Y.; Kudo, Y.; Ishimaru, N.; Funaki, M. Possible Involvement of Palmitate in Pathogenesis of Periodontitis. J. Cell. Physiol. 2015, 230, 2981–2989.
- Kim, D.J.; Rho, J.H.; Woo, B.H.; Joo, J.Y.; Lee, J.Y.; Song, J.M.; Lee, J.H.; Park, H.R. Periodontal Pathogens Modulate Lipid Flux via Fatty Acid Binding Protein 4. J. Dent. Res. 2019, 98, 1511–1520.
- Rong, X.; Albert, C.J.; Hong, C.; Duerr, M.A.; Chamberlain, B.T.; Tarling, E.J.; Ito, A.; Gao, J.; Wang, B.; Edwards, P.A.; et al. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab. 2013, 18, 685–697.
- Ito, A.; Hong, C.; Rong, X.; Zhu, X.; Tarling, E.J.; Hedde, P.N.; Gratton, E.; Parks, J.; Tontonoz, P. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife 2015, 4, e08009.
- Dos Santos, N.C.C.; Andere, N.R.M.B.; Araujo, C.F.; de Marco, A.C.; Kantarci, A.; Van Dyke, T.E.; Santamaria, M.P. Omega-3 PUFA and Aspirin as Adjuncts to Periodontal Debridement in Patients with Periodontitis and Type 2 Diabetes Mellitus. Randomized Clinical Trial. J. Periodontol. 2020, 91, 1318–1327.
- Barrea, L.; Muscogiuri, G.; Macchia, P.E.; Di Somma, C.; Falco, A.; Savanelli, M.C.; Colao, A.; Savastano, S. Mediterranean Diet and Phase Angle in a Sample of Adult Population: Results of a Pilot Study. Nutrients 2017, 9, 151.
- Varela-López, A.; Bullón, P.; Ramírez-Tortosa, C.L.; Navarro-Hortal, M.D.; Robles-Almazán, M.; Bullón, B.; Cordero, M.D.; Battino, M.; Quiles, J.L. A Diet Rich in Saturated Fat and Cholesterol Aggravates the Effect of Bacterial Lipopolysaccharide on Alveolar Bone Loss in a Rabbit Model of Periodontal Disease. Nutrients 2020, 12, 1405.
- Bastos, A.S.; Graves, D.T.; de Melo Loureiro, A.P.; Júnior, C.R.; Abdalla, D.S.P.; Faulin, T.d.E.S.; Câmara, N.O.; Andriankaja, O.M.; Orrico, S.R.P. Lipid peroxidation is associated with the severity of periodontal disease and local inflammatory markers in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2012, 97, E1353–E1362.
- Battino, M.; Bullon, P.; Wilson, M.; Newman, H. Oxidative injury and inflammatory periodontal diseases: The challenge of anti-oxidants to free radicals and reactive oxygen species. Crit. Rev. Oral Biol. Med. 1999, 10, 458–476.
- Adeshara, K.A.; Diwan, A.G.; Jagtap, T.R.; Advani, K.; Siddiqui, A.; Tupe, R.S. Relationship between plasma glycation with membrane modification, oxidative stress and expression of glucose trasporter-1 in type 2 diabetes patients with vascular complications. J. Diabetes Complicat. 2016, 31, 439–448.
- Dinicolantonio, J.J.; Mehta, V.; Onkaramurthy, N.; O’keefe, J.H. Fructose-induced inflammation and increased cortisol: A new mechanism for how sugar induces visceral adiposity. Prog. Cardiovasc. Dis. 2018, 61, 3–9.
- Saxena, A.; Tiwari, P.; Wahi, N.; Soni, A.; Bansiwal, R.C.; Kumar, A.; Sharma, B.; Punjabi, P.; Gupta, N.; Malik, B.; et al. Transcriptome profiling reveals association of peripheral adipose tissue pathology with type-2 diabetes in Asian Indians. Adipocyte 2019, 8, 125–136.
- Zhou, X.; Zhang, P.; Wang, Q.; Xia, S.; Ji, N.; Ding, Y.; Wang, Q. 25-Hydroxyvitamin D 3 Alleviates Experimental Periodontitis via Promoting Expression of Cathelicidin in Mice with Type 2 Diabetic Mellitus. J. Nutr. Sci. Vitaminol. 2018, 64, 307–315.
- Zhou, X.; Wang, Q.; Nie, L.; Zhang, P.; Zhao, P.; Yuan, Q.; Ji, N.; Ding, Y.; Wang, Q. Metformin ameliorates the NLPP3 inflammasome mediated pyroptosis by inhibiting the expression of NEK7 in diabetic periodontitis. Arch. Oral Biol. 2020, 116, 1–11.
- Zhang, D.; Jiang, Y.; Song, D.; Zhu, Y.; Zhou, C.; Dai, L.; Xu, X. Tyrosine-protein phosphatase non-receptor type 2 inhibits alveolar bone resorption in diabetic periodontitis via dephosphorylating CSF1 receptor. J. Cell. Mol. Med. 2019, 23, 6690–6699.
- Qin, X.; Hoda, M.N.; Susin, C.; Wheeler, J.N.; Marshall, B.; Perry, L.; Saad, N.; Yin, L.; Elsayed, R.; Elsalanty, M.; et al. Increased Innate Lymphoid Cells in Periodontal Tissue of the Murine Model of Periodontitis: The Role of AMP-Activated Protein Kinase and Relevance for the Human Condition. Front. Immunol. 2017, 8, 1–11.
- Zhou, X.; Zhang, P.; Wang, Q.; Ji, N.; Xia, S.; Ding, Y.; Wang, Q. Metformin ameliorates experimental diabetic periodontitis independently of mammalian target of rapamycin (mTOR) inhibition by reducing NIMA-related kinase 7 (Nek7) expression. J. Periodontol. 2019, 90, 1032–1042.
- Amir, J.; Waite, M.; Tobler, J.; Catalfamo, D.L.; Koutouzis, T.; Katz, T.; Wallet, S.M. The role of hyperglycemia in mechanisms of exacerbated inflammatory responses within the oral cavity. Cell. Immunol. 2011, 272, 45–52.
- Bala, S.V.; Appukuttan, D.; Subramaniam, S.; Prakash, P.S.G.; Cholan, P.K.; Victor, D.J. Association of Receptor for advanced glycation end products G82S polymorphism with chronic periodontitis in type II diabetic and non-diabetic South Indians. Gene 2019, 708, 30–37.
- Petrovic, S.M.; Nikolic, N.; Toljic, B.; Arambasic-Jovanovic, J.; Milicic, B.; Milicic, T.; Jotic, A.; Vidakovic, M.; Milasin, J.; Pucar, A. The association of tumor necrosis factor alpha, lymphotoxin alpha, tumor necrosis factor receptor 1 and tumor necrosis factor receptor 2 gene polymorphisms and serum levels with periodontitis and type 2 diabetes in Serbian population. Arch. Oral Biol. 2020, 120, 1–11.
- Wu, X.; Zhang, G.; Feng, X.; Li, P.; Tan, Y. Transcriptome analysis of human periodontal ligament fibroblasts exposed to Porphyromonas gingivalis LPS. Arch. Oral Biol. 2020, 110, 1–7.
- Ebersole, J.L.; Kirakodu, S.S.; Novak, M.J.; Orraca, L.; Martinez, J.G.; Cunningham, L.L.; Thomas, M.V.; Stromberg, A.; Pandruvada, S.N.; Gonzalez, O.A. Transcriptome Analysis of B Cell Immune Functions in Periodontitis: Mucosal Tissue Responses to the Oral Microbiome in Aging. Front. Immunol. 2016, 7, 1–13.
- Lin, Q.; Zhou, W.; Wang, Y.; Huang, J.; Hui, X.; Zhou, Z.; Xiao, Y. Abnormal Peripheral Neutrophil Transcriptome in Newly Diagnosed Type 2 Diabetes Patients. J. Diabetes Res. 2020, 2020, 1–10.
- Bullon, P.; Cordero, M.D.; Quiles, J.L.; Morillo, J.M.; Ramirez-Tortosa, M.D.C.; Battino, M. Mitochondrial dysfunction promoted by Porphyromonas gingivalis lipopolysaccharide as a possible link between cardiovascular disease and periodontitis. Free Radic. Biol. Med. 2011, 50, 1336–1343.
- Yan, L.; Liang-jun Yan, C. Redox imbalance stress in diabetes mellitus: Role of the polyol pathway. Anim. Model. Exp. Med. 2018, 1, 7–13.
- Quiles, J.L.; Sánchez-González, C.; Vera-Ramírez, L.; Giampieri, F.; Navarro-Hortal, M.D.; Xiao, J.; Llopis, J.; Battino, M.; Varela-López, A. Reductive Stress, Bioactive Compounds, Redox-Active Metals, and Dormant Tumor Cell Biology to Develop Redox-Based Tools for the Treatment of Cancer. ARS 2020, 33, 860–881.
- Pushparani, D.S.; Nirmala, S.; Theagarayan, P. Low serum vitamin C and zinc is associated with the development of oxidative stress in type 2 diabetes mellitus with periodontitis. Int. J. Pharm. Sci. Rev. Res. 2013, 23, 259–264.
- Smani, T.; Gallardo-Castillo, I.; Ávila-Médina, J.; Jimenez-Navarro, M.F.A.; Ordoñez, A.; Hmadcha, A. Impact of Diabetes on Cardiac and Vascular Disease: Role of Calcium Signaling. Curr. Med. Chem. 2017, 26, 4166–4177.
- Mirza, S.; Ahmed Khan, A.; Abdullah Al-Kheraif, A.; Zeb Khan, S.; Saad Shafqat, S. Efficacy of adjunctive photodynamic therapy on the clinical periodontal, HbA1c and advanced glycation end product levels among mild to moderate chronic periodontal disease patients with type 2 diabetes mellitus: A randomized controlled clinical trial. Photodiagnosis Photodyn. Ther. 2019, 28, 177–182.
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429.
- Li, J.; Guo, Y.; Chen, Y.-Y.; Liu, Q.; Chen, Y.; Tan, L.; Zhang, S.-H.; Gao, Z.-R.; Zhou, Y.-H.; Zhang, G.-Y.; et al. miR-124-3p increases in high glucose induced osteocyte-derived exosomes and regulates galectin-3 expression: A possible mechanism in bone remodeling alteration in diabetic periodontitis. FASEB J. 2020, 34, 14234–14249.
- Darrow, A.L.; Shohet, R.V. Galectin-3 deficiency exacerbates hyperglycemia and the endothelial response to diabetes. Cardiovasc. Diabetol. 2015, 14, 1–23.
- Schmidt, A.M.; Weidman, E.; Lalla, E.; Du Yan, S.; Hori, O.; Cao, R.; Brett, J.G.; Lamster, I.B. Advanced glycation endproducts (AGEs) induce oxidant stress in the gingiva: A potential mechanism underlying accelerated periodontal disease associated with diabetes. J. Periodontal Res. 1996, 31, 508–515.
- Rajeev, K.; Karthika, R.; Mythili, R.; Krishnan, V.; Nirmal, M. Role of receptors of advanced glycation end-products (RAGE) in type 2 diabetic and non-diabetic individuals with chronic periodontal disease: An immunohistochemical study. J. Investig. Clin. Dent. 2011, 2, 287–292.
- Díaz, C.M.; Bullon, B.; Ruiz-Salmerón, R.J.; Fernández-Riejos, P.; Fernández-Palacín, A.; Battino, M.; Cordero, M.D.; Quiles, J.L.; Varela-López, A.; Bullón, P. Molecular inflammation and oxidative stress are shared mechanisms involved in both myocardial infarction and periodontitis. J. Periodontal Res. 2020, 55, 519–528.
- Fazzini, F.; Lamina, C.; Raftopoulou, A.; Koller, A.; Fuchsberger, C.; Pattaro, C.; Del Greco, F.M.; Döttelmayer, P.; Fendt, L.; Fritz, J.; et al. Association of mitochondrial DNA copy number with metabolic syndrome and type 2 diabetes in 14 176 individuals. J. Intern. Med. 2021, 290, 190–202.
- Harrison, T.J.; Boles, R.G.; Johnson, D.R.; Leblond, C.; Wong, L.-J.C. Macular Pattern Retinal Dystrophy, Adult-onset Diabetes, and Deafness: A Family Study of A3243G Mitochondrial Heteroplasmy. Am. J. Ophthalmol. 1997, 124, 217–221.
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metabol. 2019, 316, E268–E285.
- Shi, Q.; Luan, Q.; Wang, X.; Cai, Y. Correlation study on mtDNA polymorphisms as potential risk factors in aggressive periodontitis by NGS. Oral Dis. 2020, 26, 401–408.
- Jiang, W.; Li, R.; Zhang, Y.; Wang, P.; Wu, T.; Lin, J.; Yu, J.; Gu, M. Mitochondrial DNA Mutations Associated with Type 2 Diabetes Mellitus in Chinese Uyghur Population. Sci. Rep. 2017, 7, 1–9.
More
Information
Subjects:
Medicine, Research & Experimental
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
884
Revisions:
2 times
(View History)
Update Date:
24 Sep 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No