 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Qing Wang | + 2117 word(s) | 2117 | 2021-09-16 06:15:11 | | | |
| 2 | Beatrix Zheng | + 979 word(s) | 3096 | 2021-09-22 12:45:11 | | |
Video Upload Options
Proteins encoded by mutant genes in cancers can be processed and presented on tumor cell surface by human leukocyte antigen (HLA) molecules, and such mutant peptides are called Neoantigens. Neoantigens are naturally existing tumor marker presented on cell surface. In clinical settings, the T-cell recognition of neoantigens is the foundation to cancer immunotherapeutics. In this article, we discussed the strategies of identifying neoantigens, followed by using phage display to create personalized cancer therapeutics -- a complete pipeline for personalized cancer treatment.
1. Introduction
The human body hosts a large amount of microbes, including archaea, bacteria, fungi, viruses, and protozoa [1][2]. Among these, phages infect bacterial hosts and can trigger the lytic replication, release of new phage particles, and new bacterial infections [3]. In addition, phages can also “collaborate” with some bacterial to kill others. The famous “kill-the-winner” model demonstrated that higher-abundance bacterial species have a greater chance of encountering virulent phages and therefore suffer more dramatically than the low abundance bacterial species, which can cause a reset to balance in abundances between different bacterial species [4]. The interaction between phage and bacterial species depends on the binding between phage surface proteins and bacteria. To utilize such features for biotechnological or therapeutic purposes, Phage Display was introduced by Smith et al. in 1985 [5]. Phage display is a process in which libraries of proteins or peptides can be displayed as fusion proteins with one of the coat proteins on the phage surface [6]. Because phage display created a simple bridge between a DNA packaged with the phage and the binding targets of the phage, it provides a powerful method for identifying the strong binders over multiple rounds of selection. Phage display can be adopted in immune library screening, where a DNA library can be first introduced into phage vectors through cloning, and the subsequent screening procedure can help identify the phages that can express antibodies or a part of an antibody that can bind with a target protein molecule [7]. Most importantly, the DNA molecule encoding this antibody or antibody fragment within the phage can be characterized for further applications. Among the many applications of phage display with immune libraries, identifying antibodies that can specifically interact with cancer cells holds the most significant clinical potential.
Cancer is one of the leading causes of human death, and it is initiated from genetic mutations that alter a normal cell’s behaviors [8][9]. Proteins encoded by mutant genes can be processed into mutation-carrying peptides and presented onto cell surface through human leukocyte antigen (HLA), and such peptides are called neoantigens [10]. Neoantigens are cancer-specific biomarkers, and they not only can distinguish cancer cells from normal but also do not induce autoimmune toxicity due to their nature of bypassing central tolerance [11]. These features make neoantigens the foundation for numerous cancer immunotherapeutic approaches, including immune checkpoint inhibitors, such as PD-1, and cancer vaccines under development [11][12]. The effectiveness of immunotherapies against cancers is often remarkable, which leads to dramatic attention to neoantigen in recent years [13][14]. With the recent development of cancer genomics readily identifying patient-specific mutations, neoantigen-based personalized therapeutics is becoming feasible [15][16]. Through phage display, we successfully developed two neoantigen-targeting personalized cancer drugs and observed phenomenal therapeutic effects [17][18].
In this review, we aim to introduce methods for building personalized cancer therapeutics through phage-eukaryotic cell interaction based on the correct identification of neoantigen as personalized therapeutic targets. We summarized a feasible technology pipeline bridging cancer genomics, immunotherapeutics as well as vaccine development through phage display to enable personalized cancer therapeutics ( Figure 1 ).
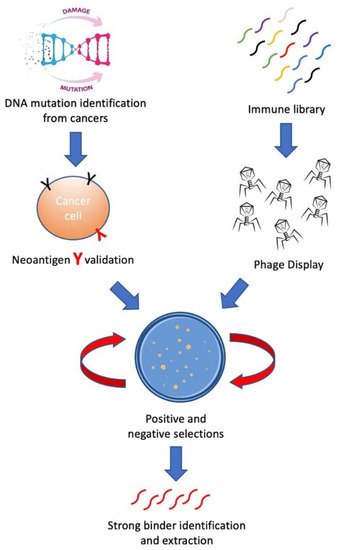
2. Neoantigen—Personalized Cancer Therapeutic Target
With the development of numerous sequencing approaches in the past two decades, the genomic information of any biological sample is readily available through highly standardized pipelines [19]. One of the most important successes in the healthcare industry in the past decade is the commercialization of next-generation sequencing (NGS) technologies into the clinical space [20]. Advances in NGS have allowed the comprehensive analysis of a cancer patient’s genome to be completed within a couple of days under 1 thousand USD nowadays as compared to taking several years with millions of dollars in the early 2000s [21]. Such fundamental changes have made cancer genome analysis now standard care for cancer patients. Patient-specific mutations can be readily identified, thus laying a solid foundation for further individualized cancer therapeutics and management.
Neoantigens represent the most personalized cancer therapeutic targets. The most important task for building up the neoantigen-based personalized cancer therapeutic method is to know the sequence and abundance of the most feasible neoantigen targets of a patient, and this critical information provides therapeutic targets for all therapeutics, including peptide vaccine, mRNA vaccine, and engineered cell therapies, etc. There are mainly three different methods to identify a neoantigen. The first method is through computation-aided prediction algorithms. With the NGS sequencing results from a tumor sample and data regarding a patient’s HLA types, possible neoantigen sequences can be predicted through numerous algorithms [22][23][24][25]. To date, there are 15 algorithms and bioinformatic platforms reported for neoantigen prediction, with the most frequently used one being NetMHC [24][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40]. Numerous other algorithms are under development, and most of them with assistance from artificial intelligence (AI) [41]. It would be a very convenient way to predict the neoantigen outright with the readily available cancer genome data; however, there are major problems in algorithm-based neoantigen predictions. First, the prediction is far from accurate. This inaccuracy comes from two directions, (1) there are only less than 5% of the predicted-to-be-presented peptides are actually presented, and (2) even for the less than 5% accurately predicted neoantigens, the affinities predicted by the algorithms are not correlated to the immunogenicity of the neoantigens presented on the cell surface [22][42][43]. Secondly, it has also been shown that only a small fraction (1–2%) of mutations are able to give rise to immunogenic neoantigens [44]. Based on these limited chances of success, it is challenging to evaluate the efficacy of a neoantigen-based therapy when only a small portion of the total dosage contains effective materials, and the treatment efficacy is expected to be at least significantly compromised [45][46].
The second method is to screen for immunogenic neoantigens that can evoke specific T-cell responses through functional analysis [47][48][49]. In this approach, tumor cells or antigen-presenting cells that are peptide-pulsed or transfected with mutation-encoding vectors are co-cultured with autologous T-cells to therefore allow the expansion of reactive T-cell clones, followed by validation procedures using tetramer staining or peptide-pulsing assays [50][51]. A major benefit in this approach is that both identifications of neoantigens and isolation of reactive T-cells that are of potential therapeutic value can be accomplished together. However, this method requires the presence of endogenous T-cell clones that can recognize the neoantigens, and such clones are either not existing or existing at an extremely low abundance level among all T-cell clonotypes. A more obvious difficulty for this approach is that it requires co-culturing of tissue cells over a relatively long period of time (~several weeks or longer); the difficulties and hefty cost coming along with the procedure made it clinically unfavorable [52].
The third method is that neoantigen peptides can be detected and quantified through mass spectrometry, which is by far the most direct way to observe neoantigens [43]. Advances in mass spectrometry have allowed for the rapid and comprehensive analysis of a peptidome sample [53][54]. However, neoantigen identification is still one of the most challenging tasks for mass spectrometry-based peptide detection [23][43]. Collaborations among well-established mass spectrometry-based proteomics labs are formed to improve method development and data sharing for neoantigen identification [55].
3. Phage-Cell Interactions and Their Therapeutic Effects
Phage represents a group of the most abundant vial entities on the planet. Based on their unique anti-bacterial feature, phage has been used for combating pathogenic bacteria in clinical treatments for over a century [56]. Due to the recent emerging issues with bacterial antibiotic resistance, phage therapy has become an important choice and has gained a lot more attention in the past decade [57]. In addition to its direct therapeutic applications, phage provides an easy linkage between the protein products and their genomes; therefore, they are widely used as a biotechnological tool to study protein-ligand interactions and to screen for therapeutic antibodies [58].
Phage therapy has been used to treat bacterial infections since the early 1900s, even before antibiotics were discovered [59]. Phage will selectively interact with bacteria by recognizing the surface proteins of the bacteria. Such selection power can allow phage to kill bacterial strains with a specific surface protein efficiently, which caused a selection pressure to the bacteria [60]. For example, phage specifically targeting efflux pumps can be selected and used to treat the bacteria that are resistant to antibiotics, and the treatment may selectively generate phage-resistant bacteria that do not have efflux pumps, and such bacteria can be further killed with antibiotics [61]. The trade-off between phage resistance and drug sensitivity would improve antimicrobial therapy and prolong the lifetime of current antibiotic therapies [62]. Such a strategy provided phage therapy a unique position in fighting against superbugs.
Phage has a unique biological feature that allows it to efficiently link the genes coding the phage and the proteins presented on the surface. Such a feature makes phage an excellent tool for antibody screening. Phage display, a technique to study the protein-ligand interaction, has been widely used in laboratories. It is one of the most effective molecular diversity techniques. Phage display is based on the fact that an encapsulated library of genotypes can be directly associated with the presentation of a library of molecules on the phage surface. Phage display has been used in a variety of applications, including epitope mapping—where a library of peptide expressing phage is used to interact with a specific antibody, therefore to pinpoint the specific epitope sequence the antibody is interacting with [63]; ligand identification for receptors—similar to antibody mapping, peptides interacting with receptors can be identified [64]; protein-protein interactions—where phage can present large proteins that are potentially interacting with a known binding partner, therefore to identify the unknown binding partners and to study the mechanism of interactions [65]; directed evolution of proteins—mutations conferring binding advantages between two proteins can be studied using a phage display library containing these variants [66]; drug discovery—peptides or ligands that can interact with drug targets can be presented through phage display [67]; and antibody screening—where a large library of antibody-displaying phage can be screened for the best antibodies that can interact with the target antigens [68].
Through phage display with multiple rounds of selection, a potential therapeutic antibody targeting neoantigen can be established. Such candidates have to be evaluated through purification and affinity measurement when they will bind with cancer cells presenting the target neoantigen [7]. Once the best clones are identified, the DNA in the phage can be extracted and used to encode antibodies that can be produced in a massive manner and adopted for cancer treatment [7]. Due to the advanced development of next-generation sequencing techniques, a phage library of stronger binders can be easily achieved [69].
4. Off-the-Shelf and Personalized Cancer Drugs Developed through Phage Display
For hotspot mutations that are shared by a large number of cancer patients, off-the-shelf antibody therapeutics targeting these neoantigens are being established by numerous lab and pharmaceutical companies [70]. The rationale behind developing such cancer therapeutics is based on the advancement of large-scale cancer genome sequencing efforts in the recent decade. As shown in Table 1 , the top 100 mutations in the human genome are responsible for close to 60% (58.23%) of all human cancers.
| DNA Change | Type | Consequences | Percentage in Cancer Patients |
|---|---|---|---|
| chr7:g.140753336A>T | Substitution | Missense BRAF V600E | 4.93% |
| chr2:g.208248388C>T | Substitution | Missense IDH1 R132H | 3.15% |
| chr12:g.25245350C>T | Substitution | Missense KRAS G12D | 2.61% |
| chr3:g.179218303G>A | Substitution | Missense PIK3CA E545K | 2.34% |
| chr3:g.179234297A>G | Substitution | Missense PIK3CA H1047R | 2.22% |
| chr12:g.25245350C>A | Substitution | Missense KRAS G12V | 2.06% |
| chr3:g.179218294G>A | Substitution | Missense PIK3CA E542K | 1.50% |
| chr17:g.7675088C>T | Substitution | Missense TP53 R175H | 1.49% |
| chr1:g.114713908T>C | Substitution | Missense NRAS Q61R | 1.44% |
| chr17:g.7674220C>T | Substitution | Missense TP53 R248Q | 1.16% |
| chr17:g.7673803G>A | Substitution | Missense TP53 R273C | 1.13% |
| chr1:g.114713909G>T | Substitution | Missense NRAS Q61K | 0.97% |
| chr12:g.25245351C>A | Substitution | Missense KRAS G12C | 0.97% |
| chr12:g.25245347C>T | Substitution | Missense KRAS G13D | 0.95% |
| chr1:g.6197725delT | Deletion | Frameshift RPL22 K15Rfs*5 | 0.93% |
| chr17:g.7673802C>T | Substitution | Missense TP53 R273H | 0.91% |
| chr17:g.7674221G>A | Substitution | Missense TP53 R248W | 0.89% |
| chr17:g.58357800delC | Deletion | Frameshift RNF43 G659Vfs*41 | 0.89% |
| chr17:g.7673776G>A | Substitution | Missense TP53 R282W | 0.83% |
| chr17:g.7674894G>A | Substitution | Stop Gained TP53 R213* | 0.72% |
| chr6:g.167003333delT | Deletion | Intron FGFR1OP | 0.69% |
| chr12:g.25227341T>G | Substitution | Missense KRAS Q61H | 0.66% |
| chr17:g.7674872T>C | Substitution | Missense TP53 Y220C | 0.65% |
| chr10:g.87965537delT | Deletion | 3 Prime UTR PTEN | 0.64% |
| chr3:g.179199088G>A | Substitution | Missense PIK3CA R88Q | 0.62% |
| chr1:g.64841314delT | Deletion | Frameshift JAK1 K860Nfs*16 | 0.57% |
| chr12:g.25245351C>G | Substitution | Missense KRAS G12R | 0.54% |
| chr17:g.7674945G>A | Substitution | Stop Gained TP53 R196* | 0.53% |
| chr17:g.20204950delA | Deletion | Frameshift SPECC1 N303Tfs*63 | 0.50% |
| chr2:g.208248389G>A | Substitution | Missense IDH1 R132C | 0.49% |
| chr10:g.87933148G>A | Substitution | Missense PTEN R130Q | 0.47% |
| chr14:g.104780214C>T | Substitution | Missense AKT1 E17K | 0.47% |
| chr9:g.21971121G>A | Substitution | Stop Gained CDKN2A R80* | 0.45% |
| chr12:g.25245350C>G | Substitution | Missense KRAS G12A | 0.45% |
| chr17:g.7674230C>T | Substitution | Missense TP53 G245S | 0.42% |
| chr10:g.87933147C>G | Substitution | Missense PTEN R130G | 0.41% |
| chr5:g.112839942C>T | Substitution | Stop Gained APC R1450* | 0.41% |
| chr17:g.7675076T>C | Substitution | Missense TP53 H179R | 0.41% |
| chr5:g.159099589delT | Deletion | 5 Prime UTR EBF1 | 0.39% |
| chr10:g.87957915C>T | Substitution | Stop Gained PTEN R233* | 0.37% |
| chr3:g.179234297A>T | Substitution | Missense PIK3CA H1047L | 0.34% |
| chr4:g.152328233G>A | Substitution | Missense FBXW7 R465C | 0.33% |
| chr7:g.140753337C>T | Substitution | Missense BRAF V600M | 0.33% |
| chr8:g.102277121delT | Deletion | Frameshift UBR5 E2121Kfs*28 | 0.33% |
| chr17:g.7670685G>A | Substitution | Stop Gained TP53 R342* | 0.33% |
| chr16:g.67611435_67611436insA | Insertion | Frameshift CTCF T204Nfs*26 | 0.33% |
| chr14:g.55684263delA | Deletion | 3 Prime UTR KTN1 | 0.32% |
| chr12:g.25245351C>T | Substitution | Missense KRAS G12S | 0.32% |
| chr17:g.7673704G>A | Substitution | Stop Gained TP53 R306* | 0.32% |
| chr4:g.1801841C>G | Substitution | Missense FGFR3 S249C | 0.32% |
| chr3:g.179203765T>A | Substitution | Missense PIK3CA N345K | 0.31% |
| chr17:g.7674953T>C | Substitution | Missense TP53 H193R | 0.30% |
| chr17:g.7675143C>A | Substitution | Missense TP53 V157F | 0.30% |
| chrX:g.77508202delA | Deletion | 3 Prime UTR ATRX | 0.30% |
| chr7:g.55191822T>G | Substitution | Missense EGFR L858R | 0.30% |
| chr17:g.39711955C>T | Substitution | Missense ERBB2 S310F | 0.30% |
| chr1:g.114716124C>G | Substitution | Missense NRAS G13R | 0.30% |
| chr19:g.3118944A>T | Substitution | Missense GNA11 Q209L | 0.30% |
| chr1:g.26779440delG | Deletion | Frameshift ARID1A D1850Tfs*33 | 0.29% |
| chr1:g.26779863C>T | Substitution | Stop Gained ARID1A R1989* | 0.29% |
| chr5:g.112840254_112840255insA | Insertion | Frameshift APC T1556Nfs*3 | 0.29% |
| chr19:g.52212718C>G | Substitution | Missense PPP2R1A P179R | 0.28% |
| chr2:g.222201320delT | Deletion | Intron PAX3 | 0.28% |
| chrX:g.40062191T>C | Substitution | Missense BCOR N1459S | 0.28% |
| chr1:g.114716126C>T | Substitution | Missense NRAS G12D | 0.27% |
| chr4:g.152328232C>T | Substitution | Missense FBXW7 R465H | 0.27% |
| chr10:g.87933147C>T | Substitution | Stop Gained PTEN R130* | 0.26% |
| chr17:g.7675994C>A | Substitution | Splice Region TP53 T125T | 0.26% |
| chr3:g.179221146G>A | Substitution | Missense PIK3CA E726K | 0.26% |
| chr17:g.7675124T>C | Substitution | Missense TP53 Y163C | 0.26% |
| chr5:g.158698822delA | Deletion | 3 Prime UTR EBF1 | 0.26% |
| chr12:g.132676598G>C | Substitution | Missense POLE P286R | 0.26% |
| chr1:g.114713908T>A | Substitution | Missense NRAS Q61L | 0.26% |
| chr12:g.4301917delT | Deletion | 3 Prime UTR CCND2 | 0.25% |
| chr12:g.49040709delG | Deletion | Frameshift KMT2D P2354Lfs*30 | 0.25% |
| chr10:g.87958013delA | Deletion | Frameshift PTEN K267Rfs*9 | 0.25% |
| chr10:g.87961042delTACT | Deletion | Frameshift PTEN T319* | 0.24% |
| chr14:g.65076348delA | Deletion | 3 Prime UTR MAX | 0.24% |
| chr9:g.77794572T>G | Substitution | Missense GNAQ Q209P | 0.24% |
| chr11:g.533874T>C | Substitution | Missense HRAS Q61R | 0.24% |
| chr3:g.179199690G>A | Substitution | Missense PIK3CA G118D | 0.24% |
| chr13:g.39343745delT | Deletion | 3 Prime UTR LHFP | 0.24% |
| chr17:g.7673802C>A | Substitution | Missense TP53 R273L | 0.24% |
| chr17:g.7675085C>A | Substitution | Missense TP53 C176F | 0.23% |
| chr10:g.121520163G>C | Substitution | Missense FGFR2 S252W | 0.23% |
| chr9:g.21971187G>A | Substitution | Stop Gained CDKN2A R58* | 0.23% |
| chr7:g.91973771delA | Deletion | Frameshift AKAP9 K39Rfs*17 | 0.23% |
| chr2:g.60458275delT | Deletion | 3 Prime UTR BCL11A | 0.23% |
| chr4:g.152326137G>C | Substitution | Missense FBXW7 R505G | 0.23% |
| chr12:g.25225628C>T | Substitution | Missense KRAS A146T | 0.23% |
| chr17:g.7674947A>G | Substitution | Missense TP53 I195T | 0.23% |
| chr5:g.112838220C>T | Substitution | Stop Gained APC R876* | 0.22% |
| chr17:g.7674957G>A | Substitution | Stop Gained TP53 Q192* | 0.22% |
| chr4:g.105240988delT | Deletion | Intron TET2 | 0.22% |
| chr17:g.7674216C>A | Substitution | Missense TP53 R249S | 0.22% |
| chr3:g.181713439delA | Deletion | 3 Prime UTR SOX2 | 0.22% |
| chr3:g.41224622C>T | Substitution | Missense CTNNB1 S37F | 0.22% |
| chr17:g.7673767C>T | Substitution | Missense TP53 E285K | 0.22% |
| chr5:g.112838934C>T | Substitution | Stop Gained APC R1114* | 0.22% |
| chr17:g.7675085C>T | Substitution | Missense TP53 C176Y | 0.22% |
| Total | 58.23% |
Such highly concentrated distribution of disease-causing mutations makes it commercially feasible for pharmaceutical companies to develop, for each mutation in Table 1 , mutation-specific neoantigen-based cancer treatment methods. It is still debating to describe the origins of tumors through monoclonal vs. multiclonal theories. However, neoantigen selection follows the same procedure regardless of monoclonality or multiclonality. The first step for neoantigen validation is to perform sequencing to identify all cancer-causing mutations, followed by ranking the mutations by allele frequency. Only the top-ranked mutations will be chosen for neoantigen validation because they represent the early events in a tumor’s development regardless of the mono- or multi- clonality.
In addition to the hotspot mutations, there are also patient-specific mutations which account for a significant part of the disease-causing mutations. To treat the disease caused by such less frequent mutations, a rapid pipeline including neoantigen validation and phage display for antibody screening can be established. It is foreseeable in the near future that, immediately after the initial diagnosis of a cancer, a patient will have a small amount of cancer tissue harvested through biopsy, and the tissue will be analyzed for personalized mutations and neoantigens. Once the neoantigen sequence and abundance are determined within a couple of days after the diagnosis, personalized cancer therapeutic agents, such as scFV, can be established through phage display within several days through rapid selection cycles. Highly personalized cancer therapy can therefore be established for each cancer patient in a timely manner.
References
- Rajagopala, S.V.; Vashee, S.; Oldfield, L.M.; Suzuki, Y.; Venter, J.C.; Telenti, A.; Nelson, K.E. The Human Microbiome and Cancer. Cancer Prev. Res. 2017, 10, 226–234.
- Cong, J.; Zhang, X. How human microbiome talks to health and disease. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1595–1601.
- Levaditi, C. Les Ultravirus: Considérés à Travers le Microscope Electronique; La Press méd: Paris, France, 1942; Volume 17, pp. 203–207.
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338.
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317.
- Ebrahimizadeh, W.; Rajabibazl, M. Bacteriophage Vehicles for Phage Display: Biology, Mechanism, and Application. Curr. Microbiol. 2014, 69, 109–120.
- Skora, A.D.; Douglass, J.; Hwang, M.S.; Tam, A.J.; Blosser, R.L.; Gabelli, S.; Cao, J.; Diaz, L.; Papadopoulos, N.; Kinzler, K.W.; et al. Generation of MANAbodies specific to HLA-restricted epitopes encoded by somatically mutated genes. Proc. Natl. Acad. Sci. USA 2015, 112, 9967–9972.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30.
- Gallia, G.L.; Zhang, M.; Ning, Y.; Haffner, M.C.; Batista, D.; Binder, Z.A.; Bishop, J.A.; Hann, C.L.; Hruban, R.H.; Ishii, M.; et al. Genomic analysis identifies frequent deletions of Dystrophin in olfactory neuroblastoma. Nat. Commun. 2018, 9, 5410.
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74.
- Heemskerk, B.; Kvistborg, P.; Schumacher, T.N.M. The cancer antigenome. EMBO J. 2012, 32, 194–203.
- Riaz, N.; Morris, L.; Havel, J.; Makarov, V.; Desrichard, A.; Chan, T.A. The role of neoantigens in response to immune checkpoint blockade. Int. Immunol. 2016, 28, 411–419.
- Larkin, J.; Sileni, V.C.; Gonzalez, R.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34.
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520.
- Ott, P.A.; Shuqiang, L.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221.
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226.
- Douglass, J.; Han-Chung Hsiue, E.; Mog, B.J.; Hwang, S.M.; Dinapoli, S.R.; Pearlamn, A.H.; Miller, M.S.; Wright, K.M.; Azurmendi, P.A.; Wang, Q.; et al. Bispecific antibodies targeting mutant RAS neoantigens. Sci. Immunol. 2021, 6, eabd5515.
- Hsiue, E.H.-C.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; Di Napoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a neoantigen derived from a common TP53 mutation. Science 2021, 371, eabc8697.
- Levy, S.E.; Myers, R.M. Advancements in Next-Generation Sequencing. Annu. Rev. Genom. Hum. Genet. 2016, 17, 95–115.
- Levy, S.E.; Boone, B.E. Next-Generation Sequencing Strategies. Cold Spring Harb. Perspect. Med. 2018, 9, a025791.
- Yohe, S.; Thyagarajan, B. Review of Clinical Next-Generation Sequencing. Arch. Pathol. Lab. Med. 2017, 141, 1544–1557.
- Bassani-Sternberg, M.; Pletscher-Frankild, S.; Jensen, L.J.; Mann, M. Mass Spectrometry of Human Leukocyte Antigen Class I Peptidomes Reveals Strong Effects of Protein Abundance and Turnover on Antigen Presentation. Mol. Cell. Proteom. 2015, 14, 658–673.
- Bassani-Sternberg, M.; Bräunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 7, 13404.
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2015, 32, 511–517.
- Zhang, H.; Lundegaard, C.; Nielsen, M. Pan-specific MHC class I predictors: A benchmark of HLA class I pan-specific prediction methods. Bioinformatics 2008, 25, 83–89.
- Kenter, G.G.; Welters, M.J.P.; Valentijn, A.R.P.M.; Lowik, M.J.G.; Der Meer, D.M.A.B.-V.; Vloon, A.P.G.; Essahsah, F.; Fathers, L.M.; Offringa, R.; Drijfhout, J.W.; et al. Vaccination against HPV-16 Oncoproteins for Vulvar Intraepithelial Neoplasia. N. Engl. J. Med. 2009, 361, 1838–1847.
- Van Poelgeest, M.I.; Welters, M.J.P.; Vermeji, R.; Stynenbisch, L.F.M.; Loof, N.M.; Berends-van der Meer, D.M.A.; Löwik, M.J.G.; Hamming, I.L.E.; van Esch, E.M.G.; Hellebrekers, B.W.J.; et al. Vaccination against Oncoproteins of HPV16 for Noninvasive Vulvar/Vaginal Lesions: Lesion Clearance Is Related to the Strength of the T-Cell Response. Clin. Cancer Res. 2016, 22, 2342–2350.
- Balachandran, V.P.; Initiative, A.P.C.G.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516.
- Kim, S.; Kim, H.S.; Kim, E.; Lee, M.G.; Shin, E.C.; Paik, S. Neopepsee: Accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immuno-genicity information. Ann. Oncol. 2018, 29, 1030–1036.
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2017, 359, 582–587.
- Hundal, J.; Carreno, B.M.; Petti, A.A.; Linette, G.P.; Griffith, O.L.; Mardis, E.R.; Griffith, M. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016, 8, 11.
- Stevanović, S.; Pasetto, A.; Helman, S.R.; Gartner, J.J.; Prickett, T.D.; Howie, B.; Robins, H.S.; Robbins, P.F.; Klebanoff, C.; Rosenberg, S.A.; et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 2017, 356, 200–205.
- Khodadoust, M.S.; Olsson, N.; Wagar, L.; Haabeth, O.A.W.; Chen, B.; Swaminathan, K.; Rawson, K.; Liu, C.L.; Steiner, D.; Lund, P.; et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature 2017, 543, 723–727.
- Liepe, J.; Marino, F.; Sidney, J.; Jeko, A.; Bunting, D.E.; Sette, A.; Kloetzel, P.M.; Stumpf, M.P.H.; Heck, A.J.R.; Mishto, M. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science 2016, 354, 354–358.
- Laumont, C.M.; Daouda, T.; Laverdure, J.-P.; Bonneil, É.; Caron-Lizotte, O.; Hardy, M.-P.; Granados, D.P.; Durette, C.; Lemieux, S.; Thibault, P.; et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat. Commun. 2016, 7, 10238.
- Abelin, J.; Keskin, D.B.; Sarkizova, S.; Hartigan, C.R.; Zhang, W.; Sidney, J.; Stevens, J.; Lane, W.; Zhang, G.L.; Eisenhaure, T.M.; et al. Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction. Immunity 2017, 46, 315–326.
- Pearson, H.; Daouda, T.; Granados, D.P.; Durette, C.; Bonneil, E.; Courcelles, M.; Rodenbrock, A.; Laverdure, J.-P.; Côté, C.; Mader, S.; et al. MHC class I–associated peptides derive from selective regions of the human genome. J. Clin. Investig. 2016, 126, 4690–4701.
- Jørgensen, K.W.; Rasmussen, M.; Buus, S.; Nielsen, M. NetMHCstab- predicting stability of peptide-MHC-I complexes; impacts for cytotoxic T lymphocyte epitope discovery. Immunology 2013, 141, 18–26.
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Brunak, S.; Lund, O.; Nielsen, M. An integrative approach to CTL epitope prediction: A combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur. J. Immunol. 2005, 35, 2295–2303.
- Singh-Jasuja, H.; Emmerich, N.P.N.; Rammensee, H.-G. The Tüingen approach: Identification, selection, and validation of tumor-associated HLA peptides for cancer therapy. Cancer Immunol. Immunother. 2004, 53, 187–195.
- Wu, J.; Wang, W.; Zhang, J.; Zhou, B.; Zhao, W.; Su, Z.; Gu, X.; Wu, J.; Zhou, Z.; Chen, S.; et al. DeepHLApan: A Deep Learning Approach for Neoantigen Prediction Considering Both HLA-Peptide Binding and Immunogenicity. Front. Immunol. 2019, 10, 2559.
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.J.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576.
- Wang, Q.; Douglass, J.; Hwang, M.S.; Hsiue, E.H.-C.; Mog, B.J.; Zhang, M.; Papadopoulos, N.; Kinzler, K.W.; Zhou, S.; Vogelstein, B. Direct Detection and Quantification of Neoantigens. Cancer Immunol. Res. 2019, 7, 1748–1754.
- Tran, E.; Ahmadzadeh, M.; Lu, Y.-C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390.
- Anonymous. The problem with neoantigen prediction. Nat. Biotechnol. 2017, 35, 97.
- Vitiello, A.; Zanetti, M. Neoantigen prediction and the need for validation. Nat. Biotechnol. 2017, 35, 815–817.
- Hadrup, S.R.; Bakker, A.; Shu, C.J.; Andersen, R.S.; Van Veluw, J.; Hombrink, P.; Castermans, E.; Straten, P.T.; Blank, C.; Haanen, J.B.; et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat. Methods 2009, 6, 520–526.
- Bentzen, A.K. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA bar-codes. Nat. Biotechnol. 2016, 34, 1037–1045.
- Bentzen, A.K.; Hadrup, S.R. Evolution of MHC-based technologies used for detection of antigen-responsive T cells. Cancer Immunol. Immunother. 2017, 66, 657–666.
- Lu, Y.-C.; Yao, X.; Crystal, J.S.; Li, Y.F.; El-Gamil, M.; Gross, C.; Davis, L.; Dudley, M.E.; Yang, J.C.; Samuels, Y.; et al. Efficient Identification of Mutated Cancer Antigens Recognized by T Cells Associated with Durable Tumor Regressions. Clin. Cancer Res. 2014, 20, 3401–3410.
- Danilova, L.; Anagnostou, V.; Caushi, J.X.; Sidhom, J.-W.; Guo, H.; Chan, H.Y.; Suri, P.; Tam, A.J.; Zhang, J.; El Asmar, M.; et al. The Mutation-Associated Neoantigen Functional Expansion of Specific T Cells (MANAFEST) Assay: A Sensitive Platform for Monitoring Antitumor Immunity. Cancer Immunol. Res. 2018, 6, 888–899.
- Kato, T.; Matsuda, T.; Ikeda, Y.; Park, J.-H.; Leisegang, M.; Yoshimura, S.; Hikichi, T.; Harada, M.; Zewde, M.G.; Sato, S.; et al. Effective screening of T cells recognizing neoantigens and construction of T-cell receptor-engineered T cells. Oncotarget 2018, 9, 11009–11019.
- Kim, M.-S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581.
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587.
- Caron, E.; Aebersold, R.; Banaei-Esfahani, A.; Chong, C.; Bassani-Sternberg, M. A Case for a Human Immuno-Peptidome Project Consortium. Immunity 2017, 47, 203–208.
- Shasha, S.M.; Sharon, N.; Inbar, M. [Bacteriophages as antibacterial agents]. Harefuah 2004, 143, 121–125.
- Altamirano, F.L.G.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev. 2019, 32.
- Tan, Y.; Tian, T.; Liu, W.; Zhu, Z.; Yang, C.J. Advance in phage display technology for bioanalysis. Biotechnol. J. 2016, 11, 732–745.
- Summers, W.C. The strange history of phage therapy. Bacteriophage 2012, 2, 130–133.
- Labrie, S.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Genet. 2010, 8, 317–327.
- Liu, C.G.; Green, S.I.; Min, L.; Clark, J.R.; Salazar, K.; Terwilliger, A.L.; Kaplan, H.B.; Trautner, B.W.; Ramig, R.F.; Maresso, A.W. Phage-Antibiotic Synergy Is Driven by a Unique Combination of Antibacterial Mechanism of Action and Stoichiometry. MBio 2020, 11.
- Gurney, J.; Pradier, L.; Griffin, J.S.; Gougat-Barbera, C.; Chan, B.K.; Turner, E.P.; Kaltz, O.; Hochberg, E.M. Phage steering of antibiotic-resistance evolution in the bacterial pathogen, Pseudomonas aeruginosa. Evol. Med. Public Health 2020, 2020, 148–157.
- Moreira, G.M.S.G.; Fühner, V.; Hust, M. Epitope Mapping by Phage Display. J. Immunol. Methods 2017, 1701, 497–518.
- Hartley, O. The use of phage display in the study of receptors and their ligands. J. Recept. Signal Transduct. 2002, 22, 373–392.
- Kushwaha, R.; Payne, C.M.; Downie, A.B. Uses of phage display in agriculture: A review of food-related pro-tein-protein interactions discovered by biopanning over diverse baits. Comput. Math. Methods Med. 2013, 2013, 653759.
- Rojas, G.; Carmenate, T.; Santo-Tomás, J.F.; Valiente, P.A.; Becker, M.; Riverón, A.P.; Tundidor, Y.; Ortiz, Y.; De Cossio-Diaz, J.F.; Graca, L.; et al. Directed evolution of super-secreted variants from phage-displayed human Interleukin-2. Sci. Rep. 2019, 9, 800.
- Mimmi, S.; Maisano, D.; Quinto, I.; Iaccino, E. Phage Display: An Overview in Context to Drug Discovery. Trends Pharmacol. Sci. 2019, 40, 87–91.
- Jara-Acevedo, R.; Díez, P.; González-González, M.; Dégano, R.M.; Ibarrola, N.; Góngora, R.; Orfao, A.; Fuentes, M. Screening Phage-Display Antibody Libraries Using Protein Arrays. Methods Mol. Biol. 2017, 1701, 365–380.
- Rouet, R.; Jackson, K.J.L.; Langley, D.B.; Christ, D. Next-Generation Sequencing of Antibody Display Repertoires. Front. Immunol. 2018, 9, 118.
- Zhao, W.; Wu, J.; Chen, S.; Zhou, Z. Shared neoantigens: Ideal targets for off-the-shelf cancer immunotherapy. Pharmacogenomics 2020, 21, 637–645.

