 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Michael D Faulx | + 1810 word(s) | 1810 | 2021-09-04 13:31:04 | | | |
| 2 | Conner Chen | Meta information modification | 1810 | 2021-09-23 02:37:38 | | |
Video Upload Options
Sleep apnea is a highly prevalent disorder among patients with all forms of cardiovascular disease. Decades of data from several large prospective patient registries have revealed that sleep apnea—in particular, obstructive sleep apnea (OSA)—is practically endemic in cardiology clinics and cardiac inpatient wards across the globe. OSA has been closely associated with prevalent and incident hypertension, ischemic heart disease, heart failure, stroke, and all forms of cardiac rhythm disturbance. Additionally, central sleep apnea (CSA) or combined OSA and CSA often affects patients with heart failure and stroke. Sleep apnea and cardiovascular disease are so intertwined with respect to their epidemiology and shared pathophysiology that one can think of them as being two components of a global, multi-system metabolic syndrome driven largely by obesity.
1. The Epidemiology of Obstructive Sleep Apnea
OSA is a global health crisis that parallels the global obesity epidemic. Obesity and OSA are associated to the extent that it is credible to think of OSA as a consequence of obesity in a majority of cases, although there are certainly patients with OSA who are not obese. In the United States, OSA affects 17% of adult women and 34% of adult men, and incident cases are on the rise [1]. Across the world, OSA prevalence rates vary, but share the trend of rising on every continent [2]. OSA is often associated with features of the metabolic syndrome or “Syndrome X”, including insulin resistance, dyslipidemia, hypertension, and central adiposity, so often so that some authors have proposed the adoption of a “Syndrome Z” to account for the frequent presence of OSA [3]. This association with the metabolic syndrome and its attendant effects on inflammation, oxidative stress, and endothelial dysfunction likely accounts for a large portion of the association between OSA and cardiovascular disease [4].
OSA is often symptomatic, with its principal symptom being excessive daytime sleepiness or fatigue. Historical features that are strongly suggestive of OSA include loud snoring and witnessed apneas or gasping for air during sleep. This element of the history often requires an interview with the patient’s bed partner for confirmation. Other symptomatic manifestations of OSA include difficult concentration, declining work performance, depressed mood, and a heightened risk for motor vehicle accidents. There are a number of valid and simple screening tools that can be easily applied during a patient interview to predict the presence of OSA with fair accuracy. Of these, the STOP-BANG questionnaire (Table 1) appears to have the best sensitivity and specificity for the detection of OSA [5][6][7]. Although many patients with OSA do not volunteer that they are symptomatic, screening for symptoms can nonetheless be helpful. The Epworth sleepiness scale (ESS), an eight-item questionnaire administered during a clinical encounter (Table 2), can prove useful in establishing whether significant OSA symptoms are present [8]. While it is not solely specific to sleepiness caused by sleep apnea, the ESS scale has been well validated in the OSA population and is a reliable gauge for symptom severity. This matters because the presence of subjective and objective sleepiness correlates with greater expression of pro-inflammatory biomarkers and a greater overall risk for adverse cardiac events than the absence of OSA symptoms [9]. The presence of symptoms also justifies OSA treatment, irrespective of any interest in cardiac risk mitigation.
Table 1. The STOP-BANG questionnaire and its accuracy in detecting moderate or severe sleep apnea (AHI ≥15/hour). Score one point for each finding.
| Snoring | Typically loud and disruptive | |||
| Tiredness | Tired, fatigued, or sleepy during the day | |||
| Observed apnea | Often observed by bed partner | |||
| Pressure | History of hypertension treatment | |||
| BMI | BMI > 35 kg/m2 | |||
| Age | >50 years | |||
| Neck circumference | >40 cm | |||
| Gender | Male | |||
| STOP BANG Score | Sensitivity | Specificity | PPV | NPV |
| 1 | 100 | 1 | 67 | 100 |
| 2 | 99 | 10 | 68 | 79 |
| 3 | 94 | 32 | 73 | 74 |
| 4 | 81 | 51 | 76 | 58 |
| 5 | 60 | 72 | 80 | 48 |
| 6 | 35 | 89 | 86 | 42 |
| 7 | 14 | 96 | 88 | 37 |
| 8 | 3 | 100 | 95 | 35 |
Table 2. The Epworth Sleepiness Scale (ESS) and its relationship to OSAS risk. Each question is scored from 0 to 3. ESS score range is from 0–24.
| Activity | Likelihood of Dozing 0 = Never, 1 = Slight, 2 = Moderate, 3 = High |
||
|---|---|---|---|
| Sitting and reading | |||
| Watching television | |||
| Sitting inactively in a public place | |||
| As a car passenger for one uninterrupted hour | |||
| Lying down in the afternoon when able | |||
| Sitting and talking to someone | |||
| Sitting quietly after lunch with no alcohol | |||
| In a car, while stopped for a few minutes in traffic | |||
| Mean RDI | Mean ESS | ESS Range | Interpretation |
| 8.8 ± 2.3 | 9.5 ± 3.3 | 1–9 | No or little OSAS risk |
| 21.1 ± 4.0 | 11.5 ± 4.2 | 10–15 | Moderate OSAS risk |
| 49.5 ± 9.6 | 16.0 ± 4.4 | 16–24 | High OSAS risk |
RDI, respiratory disturbance index; OSAS, obstructive sleep apnea syndrome. The table was adapted from reference [10].
When OSA is strongly suspected after screening, the diagnosis is typically confirmed or excluded with an attended, laboratory-based polysomnogram (PSG) or a home sleep apnea test (HSAT). PSG is considered the gold standard for the diagnosis of sleep disorders owing to its multi-channel data acquisition, which includes brainwave activity and cardiac telemetry to allow for sleep staging, arousal assessment, and assessment of heart rate variability. Major disadvantages of PSG include its limited availability despite rising demand and lack of access in the setting of the ongoing coronavirus disease 2019 (COVID-19) pandemic [10]. In contrast, HSAT offers a simpler dataset that includes continuous oximetry and airflow assessment. The device is worn in the patient’s home and is often more readily available than PSG. HSAT is most appropriate for patients with few medical comorbidities in whom there is a high index of suspicion for OSA, rather than central or mixed apnea. There are even algorithms that allow for the assessment of heart rate variability based on data obtained from continuous oximetry, a feature that may prove useful in the prediction of incident rhythm disorders, such as atrial fibrillation [11].
OSA severity may be assessed in several ways. The most commonly reported metric of OSA severity is the apnea hypopnea index (AHI), which measures the number of times that a patient stops breathing (apnea) or experiences a significant reduction in airflow (hypopnea) per hour of sleep time. An apnea is defined as a lack of an air flow for at least 10 s with an associated oxygen desaturation of at least 4%. Hypopnea is defined as a 50% or greater reduction in airflow for at least 10 s with an associated oxygen desaturation of at least 4%. The AHI is easy to reproduce and is, without question, the most widely reported OSA severity metric in clinical trials. However, the AHI may underrepresent OSA severity when viewed in isolation, and there are data to support focusing more on indices of oxygen desaturation as a gauge of OSA severity [12]. Recent studies have suggested that measures of oxygen desaturation, such as the percentage of sleep time spent with an oxygenation saturation below 90% (T90) or 88% (T88) or the lowest saturation achieved during sleep, may better predict adverse cardiac events than the AHI [13].
2. Obstructive Sleep Apnea and Cardiac Arrhythmogenesis
OSA impacts the development of cardiac arrhythmias through direct and indirect mechanisms (Figure 1). The direct effects of OSA on arrhythmia development include the acute physiologic changes that occur as a consequence of airway collapse during sleep, including the development of hypoxemia and hypercapnia [14], changes in sympathetic and parasympathetic tone [15], and fluctuations in thoracic pressure [16]. Indirectly, OSA alters the structure of the heart and is a risk factor for the development of structural heart disease. The indirect effects include the development of cardiovascular disease, including hypertension [17][18], heart failure, and coronary artery disease [19], which form the underlying substrate for arrhythmia development. While atrial fibrillation is the arrhythmia most commonly associated with OSA [20], there is evidence linking OSA to the development of arrhythmias at the level of the sinus node [21], atrial arrhythmias [22], ventricular arrhythmias, and sudden cardiac death [23]. In this section, we will review the pathophysiologic impact of sleep apnea on cardiac arrhythmia and explore the relationship with each disease entity in turn.
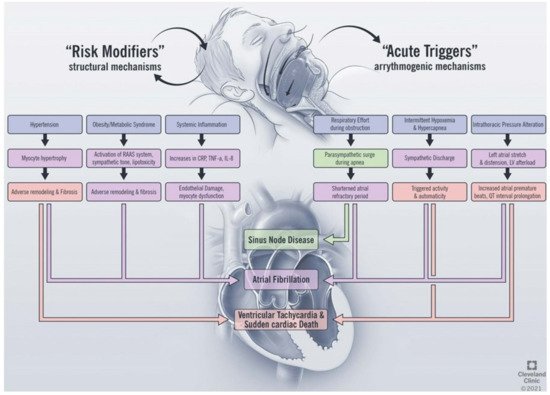
Figure 1. Proposed mechanisms linking obstructive sleep apnea and cardiac arrhythmias. RAAS, renin angiotensin aldosterone system; CRP, c-reactive protein; TNF, tumor necrosis factor; IL, interleukin; LV, left ventricle.
The basis of arrhythmogenesis includes changes in myocardial automaticity, triggered activity, and reentrant mechanisms [24]. Abnormal automaticity refers to the formation of cardiac impulses in normally quiescent cardiac cells and is controlled by multiple factors, including sympathetic and parasympathetic tone, acid–base status, and electrolyte disturbances at the membrane and sub-membrane levels [25]. OSA causes repetitive, cyclical changes in sympathetic tone. During apneic events, increased vagal tone causes bradycardia followed by sympathetic discharge as a result of hypoxemia and hypercapnia. Increased vagal tone has been shown to shorten the effective refractory period of the atrium in porcine models of AF and to lead to easier inducibility of AF [26]. The following sympathetic discharge, in turn, promotes increased arrhythmia formation due to beta-adrenergic stimulation [16][27] The repetitive hypoxemia is also thought to increase reactive oxygen species and alter potassium regulation during sleep, which affects the automaticity of cardiac tissue [28]. OSA has also been shown to decrease the atrial effective refractory period (ERP) in canine models, thus leaving the atria more vulnerable to automatic depolarization and ectopy during periods of sleep-disordered breathing [29]. Triggered activity refers to spontaneous depolarizations that are able to cross the membrane potential required to trigger an action potential. These individual extrasystoles can precipitate tachyarrhythmias in both the atrial and ventricular chambers [29]. Well-established causes of triggered activity include hypoxemia, acidemia, and increased sympathetic tone, all of which occur during the repetitive cycles of apnea that characterize OSA. Re-entrant mechanisms are postulated to arise from heterogenous myocardial conduction as a result of abnormal cardiac remodeling in the setting of structural heart disease that accompanies OSA [14].
The mechanistic link between OSA and heart failure is complex and likely bidirectional, with each entity contributing to the other [30]. Obstructive apnea and hypopnea are associated with respiratory efforts against the collapsed upper airway, with associated changes in intrathoracic pressure as high as 60 to 80 mmHg [31] These repetitive, acute swings have a significant impact on cardiac preload and afterload. Simulation of OSA by means of the Mueller maneuver, which involves breathing in against a forced resistance by means of a nose clip and mouthpiece with a 21 G needle, was shown to reproduce changes in intrathoracic pressure in healthy human subjects [29]. This experiment showed that the effect of these intrathoracic pressure swings includes increases in left ventricle (LV) end-systolic volumes, decreased cardiac performance, and abrupt swings in left atrial volumes due to mural stress on the more pliable left atrial wall. Likewise, in patients undergoing cardiac catheterization with measurement of aortic and left ventricular pressures, negative intrathoracic pressures by means of the Mueller maneuver caused increases in LV contraction load as well as an increase in the LV relaxation coefficient (tau) [32][33]. These pathophysiologic changes play a possible role in the observation that severe OSA is associated with ventricular diastolic dysfunction in a dose-dependent fashion [33]. The sum of these interactions is that OSA predisposes one to the development of structural heart disease and heart failure, and the development of these disease states, in turn, predisposes one to and perpetuates the development of OSA.
References
- Peppard, P.E.; Young, T.; Barnet, J.H.; Palta, M.; Hagen, E.W.; Hla, K.M. Increased Prevalence of Sleep-Disordered Breathing in Adults. Am. J. Epidemiology 2013, 177, 1006–1014.
- Senaratna, C.V.; Perret, J.L.; Lodge, C.J.; Lowe, A.J.; Campbell, B.E.; Matheson, M.C.; Hamilton, G.S.; Dharmage, S.C. Prevalence of obstructive sleep apnea in the general population: A systematic review. Sleep Med. Rev. 2016, 34, 70–81.
- Jehan, S.; Myers, A.K.; Zizi, F.; Perumal, S.R.P.; Louis, G.J.; McFarlane, S.I. Obesity, obstructive sleep apnea and type diabetes mellitus: Epidemiology and pathophysiologic insights. Sleep Med. Disord. Int. J. 2018, 2, 52–58.
- Jean-Louis, G.; Zizi, F.; Clark, L.T.; Brown, C.D.; McFarlane, S.I. Obstructive Sleep Apnea and Cardiovascular Disease: Role of the Metabolic Syndrome and Its Components. J. Clin. Sleep Med. 2008, 4, 261–272.
- Chung, F.; Abdullah, H.R.; Liao, P. STOP-Bang Questionnaire: A Practical Approach to Screen for Obstructive Sleep Apnea. Chest 2016, 149, 631–638.
- Chiu, H.-Y.; Chen, P.-Y.; Chuang, L.-P.; Chen, N.-H.; Tu, Y.-K.; Hsieh, Y.-J.; Wang, Y.-C.; Guilleminault, C. Diagnostic accuracy of the Berlin questionnaire, STOP-BANG, STOP, and Epworth sleepiness scale in detecting obstructive sleep apnea: A bivariate meta-analysis. Sleep Med. Rev. 2017, 6, 57–70.
- Nagappa, M.; Liao, P.; Wong, J.; Auckley, D.; Ramachandran, S.K.; Memtsoudis, S.G.; Mokhlesi, B.; Chung, F. Validation of the STOP-Bang Questionnaire as a Screening Tool for Obstructive Sleep Apnea among Different Populations: A Systematic Review and Meta-Analysis. PLOS ONE 2015, 10, e0143697.
- Johns, M.W. Daytime sleepiness, snoring, and obstructive sleep apnea. The Epworth Sleepiness Scale. Chest 1993, 103, 30–36.
- Mehra, R.; Wang, L.; Andrews, N.; Tang, W.W.; Young, J.B.; Javaheri, S.; Foldvary-Schaefer, N. Dissociation of Objective and Subjective Daytime Sleepiness and Biomarkers of Systemic Inflammation in Sleep-Disordered Breathing and Systolic Heart Failure. J. Clin. Sleep Med. 2017, 13, 1411–1422.
- Johnson, K.G.; Sullivan, S.S.; Rastegar, A.N.V.; Gurubhagavatula, I. The impact of the COVID-19 pandemic on sleep medicine practices. J. Clin. Sleep Med. 2021, 7, 79–87.
- Blanchard, M.; Gervès-Pinquié, C.; Feuilloy, M.; Le Vaillant, M.; Trzepizur, W.; Meslier, N.; Paris, A.; Pigeanne, T.; Racineux, J.-L.; Balusson, F.; et al. Association of Nocturnal Hypoxemia and Pulse Rate Variability with Incident Atrial Fibrillation in Patients Investigated for Obstructive Sleep Apnea. Ann. Am. Thorac. Soc. 2021, 18, 1043–1051.
- Kainulainen, S.; Töyräs, J.; Oksenberg, A.; Korkalainen, H.; Sefa, S.; Kulkas, A.; Leppänen, T. Severity of Desaturations Reflects OSA-Related Daytime Sleepiness Better Than AHI. J. Clin. Sleep Med. 2019, 15, 1135–1142.
- Xie, J.; Kuniyoshi, F.H.S.; Covassin, N.; Singh, P.; Gami, A.S.; Wang, S.; Chahal, C.A.A.; Wei, Y.; Somers, V.K. Nocturnal Hypoxemia Due to Obstructive Sleep Apnea Is an Independent Predictor of Poor Prognosis After Myocardial Infarction. J. Am. Hear. Assoc. 2016, 5.
- May, A.M.; van Wagoner, D.R.; Mehra, R. OSA. Cardiac Arrhythmogenesis: Mechanistic Insights. Chest 2017, 51, 225–241.
- Leung, R.S. Sleep-Disordered Breathing: Autonomic Mechanisms and Arrhythmias. Prog. Cardiovasc. Dis. 2009, 51, 324–338.
- Drager, L.F.; Silva, L.D.; Diniz, P.M.; Bortolotto, L.A.; Pedrosa, R.P.; Couto, R.B.; Marcondes, B.; Giorgi, D.M.A.; Filho, G.L.; Krieger, E.M. Obstructive sleep apnea, hypertension, and their interaction on arterial stiffness and heart remodeling. Chest 2007, 31, 1379–1386.
- Guillot, M.; Sforza, E.; Crawford, E.A.; Maudoux, D.; Martin, M.S.; Barthélémy, J.-C.; Roche, F. Association between severe obstructive sleep apnea and incident arterial hypertension in the older people population. Sleep Med. 2013, 838–842.
- Marin, J.M.; Agusti, A.; Villar, I.; Forner, M.; Nieto, D.; Carrizo, S.J.; Barbé, F.; Vicente, E.; Wei, Y.; Nieto, F.J.; et al. Association Between Treated and Untreated Obstructive Sleep Apnea and Risk of Hypertension. JAMA 2012, 07, 2169–2176.
- Shahar, E.; Whitney, C.W.; Redline, S.; Lee, E.T.; Newman, A.B.; Nieto, F.J.; O’Connor, G.T.; Boland, L.L.; Schwartz, J.E.; Samet, J.M. Sleep-disordered Breathing and Cardiovascular Disease. Am. J. Respir. Crit. Care Med. 2001, 163, 19–25.
- Chung, M.K.; Eckhardt, L.L.; Chen, L.Y.; Ahmed, H.M.; Gopinathannair, R.; Joglar, J.A.; Noseworthy, P.A.; Pack, Q.R.; Sanders, P.; Trulock, K.M. Lifestyle and Risk Factor Modification for Reduction of Atrial Fibrillation: A Scientific Statement From the American Heart Association. Circulation 2020, 41, e750–e772.
- Almor, J.M.; López, J.J.; Casteigt, B.; Conejos, J.; Valles, E.; Farré, N.; Flor, M.F. Prevalence of obstructive sleep apnea syndrome in patients with sick sinus syndrome. Rev. Esp. Cardiol. 2006, 9, 28–32.
- Hoffstein, V.; Mateika, S. Cardiac arrhythmias, snoring, and sleep apnea. Chest 1994, 106, 466–471.
- Gami, A.S.; Howard, D.E.; Olson, E.J.; Somers, V.K. Day-night pattern of sudden death in obstructive sleep apnea. N. Engl. J. Med. 2005, 52, 1206–1214.
- Mann, D.; Zipes, D.; Libby, P.; Bonow, R. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine, 11th ed.; Elsevier/Saunders: Philadelphia, PA, USA, 2015.
- Vetulli, H.M.; Elizari, M.V.; Naccarelli, G.V.; Gonzalez, M.D. Cardiac automaticity: Basic concepts and clinical observations. J. Interv. Card. Electrophysiol. 2018, 52, 263–270.
- Linz, D.; Schotten, U.; Neuberger, H.-R.; Böhm, M.; Wirth, K. Negative tracheal pressure during obstructive respiratory events promotes atrial fibrillation by vagal activation. Hear. Rhythm. 2011, 8, 1436–1443.
- Chadda, K.R.; Fazmin, I.T.; Ahmad, S.; Valli, H.; E Edling, C.; Huang, C.L.-H.; Jeevaratnam, K. Arrhythmogenic mechanisms of obstructive sleep apnea in heart failure patients. Sleep 2018, 41.
- Lu, Z.; Nie, L.; He, B.; Yu, L.; Salim, M.; Huang, B.; Cui, B.; He, W.; Wu, W.; Jiang, H. Increase in vulnerability of atrial fibrillation in an acute intermittent hypoxia model: Importance of autonomic imbalance. Auton. Neurosci. 2013, 177, 148–153.
- Tse, G. Mechanisms of cardiac arrhythmias. J. Arrhythmia 2015, 32, 75–81.
- Selim, B.J.; Ramar, K. Management of Sleep Apnea Syndromes in Heart Failure. Sleep Med. Clin. 2017, 12, 107–121.
- Orban, M.; Bruce, C.J.; Pressman, G.S.; Leinveber, P.; Romero-Corral, A.; Korinek, J.; Konecny, T.; Villarraga, H.R.; Kara, T.; Caples, S.M.; et al. Dynamic Changes of Left Ventricular Performance and Left Atrial Volume Induced by the Mueller Maneuver in Healthy Young Adults and Implications for Obstructive Sleep Apnea, Atrial Fibrillation, and Heart Failure. Am. J. Cardiol. 2008, 102, 1557–1561.
- Fung, J.W.; Li, T.S.; Choy, D.K.; Yip, G.W.; Ko, F.W.; Sanderson, J.E.; Hui, D.S. Severe Obstructive Sleep Apnea Is Associated With Left Ventricular Diastolic Dysfunction. Chest 2002, 121, 422–429.
- Virolainen, J.; Ventila, M.; Turto, H.; Kupari, M. Effect of negative intrathoracic pressure on left ventricular pressure dynamics and relaxation. J. Appl. Physiol. 1995, 79, 455–460.

