 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Manisha Jalan | + 1985 word(s) | 1985 | 2021-08-25 08:35:22 | | | |
| 2 | Vivi Li | Meta information modification | 1985 | 2021-09-14 05:21:34 | | |
Video Upload Options
The maintenance of genome integrity is critical for cell survival. Homologous recombination (HR) is considered the major error-free repair pathway in combatting endogenously generated double-stranded lesions in DNA. Nevertheless, a number of alternative repair pathways have been described as protectors of genome stability, especially in HR-deficient cells. One of the factors that appears to have a role in many of these pathways is human RAD52, a DNA repair protein that was previously considered to be dispensable due to a lack of an observable phenotype in knock-out mice. In later studies, RAD52 deficiency has been shown to be synthetically lethal with defects in BRCA genes, making RAD52 an attractive therapeutic target, particularly in the context of BRCA-deficient tumors.
1. Double-Strand Breaks and RAD52
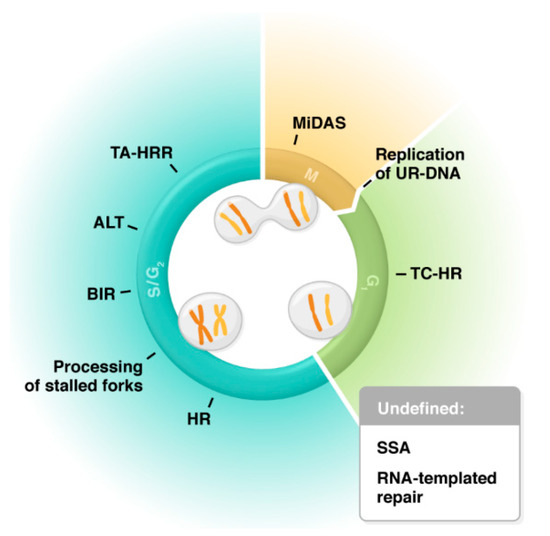
2. Biochemistry and Activity of RAD52
3. RAD52 as a Target for Cancer Therapy
References
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64.
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Boil. Chem. 2018, 293, 10502–10511.
- Lok, B.H.; Powell, S.N. Molecular pathways: Understanding the role of Rad52 in homologous recombination for therapeutic advancement. Clin. Cancer Res. 2012, 18, 6400–6406.
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Boil. 2010, 11, 196–207.
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Boil. 2019.
- Zhao, W.; Steinfeld, J.B.; Liang, F.; Chen, X.; Maranon, D.G.; Ma, C.J.; Kwon, Y.; Rao, T.; Wang, W.; Sheng, C.; et al. BRCA1–BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 2017, 550, 360–365.
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510.
- Saleh-Gohari, N.; Bryant, H.E.; Schultz, N.; Parker, K.M.; Cassel, T.N.; Helleday, T. Spontaneous Homologous Recombination Is Induced by Collapsed Replication Forks That Are Caused by Endogenous DNA Single-Strand Breaks. Mol. Cell. Boil. 2005, 25, 7158–7169.
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single Strand Annealing and its role in genome maintenance. Trends Genet. 2016, 32, 566–575.
- Bétermier, M.; Bertrand, P.; Lopez, B.S. Is Non-Homologous End-Joining Really an Inherently Error-Prone Process? PLoS Genet. 2014, 10, e1004086.
- Stark, J.M.; Pierce, A.J.; Oh, J.; Pastink, A.; Jasin, M. Genetic Steps of Mammalian Homologous Repair with Distinct Mutagenic Consequences. Mol. Cell. Boil. 2004, 24, 9305–9316.
- Mortensen, U.H.; Bendixen, C.; Sunjevaric, I.; Rothstein, R. DNA strand annealing is promoted by the yeast Rad52 protein. Proc. Natl. Acad. Sci. USA 2002, 93, 10729–10734.
- Reddy, G.; Golub, E.I.; Radding, C.M. Human Rad52 protein promotes single-strand DNA annealing followed by branch migration. Mutat. Res. Mol. Mech. Mutagen. 1997, 377, 53–59.
- Symington, L.S. Role of RAD52 Epistasis Group Genes in Homologous Recombination and Double-Strand Break Repair. Microbiol. Mol. Boil. Rev. 2002, 66, 630–670.
- Kan, Y.; Batada, N.N.; Hendrickson, E.A. Human somatic cells deficient for RAD52 are impaired for viral integration and compromised for most aspects of homology-directed repair. DNA Repair 2017, 55, 64–75.
- Hanamshet, K.; Mazina, O.M.; Mazin, A.V. Reappearance from Obscurity: Mammalian Rad52 in Homologous Recombination. Genes 2016, 7, 63.
- Game, J.; Mortimer, R. A genetic study of X-ray sensitive mutants in yeast. Mutat. Res. Toxicol. 1974, 24, 281–292.
- Sugiyama, T.; New, J.H.; Kowalczykowski, S.C. DNA annealing by Rad52 Protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc. Natl. Acad. Sci. USA 1998, 95, 6049–6054.
- Rijkers, T.; Ouweland, J.V.D.; Morolli, B.; Rolink, A.G.; Baarends, W.M.; Van Sloun, P.P.H.; Lohman, P.H.M.; Pastink, A. Targeted Inactivation of Mouse RAD52 Reduces Homologous Recombination but Not Resistance to Ionizing Radiation. Mol. Cell. Boil. 1998, 18, 6423–6429.
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691.
- Lok, B.H.; Carley, A.C.; Tchang, B.; Powell, S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 2013, 32, 3552–3558.
- Ma, C.J.; Kwon, Y.; Sung, P.; Greene, E.C. Human RAD52 interactions with replication protein A and the RAD51 presynaptic complex. J. Boil. Chem. 2017, 292, 11702–11713.
- Liu, Y.; Li, M.-J.; Lee, E.Y.-H.; Maizels, N. Localization and dynamic relocalization of mammalian Rad52 during the cell cycle and in response to DNA damage. Curr. Boil. 1999, 9, 975–978.
- Murfuni, I.; Basile, G.; Subramanyam, S.; Malacaria, E.; Bignami, M.; Spies, M.; Franchitto, A.; Pichierri, P. Survival of the Replication Checkpoint Deficient Cells Requires MUS81-RAD52 Function. PLoS Genet. 2013, 9, e1003910.
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126.
- Sotiriou, S.K.; Kamileri, I.; Lugli, N.; Evangelou, K.; Da-Ré, C.; Huber, F.; Padayachy, L.; Tardy, S.; Nicati, N.L.; Barriot, S.; et al. Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol. Cell 2016, 64, 1127–1134.
- Malacaria, E.; Pugliese, G.M.; Honda, M.; Marabitti, V.; Aiello, F.A.; Spies, M.; Franchitto, A.; Pichierri, P. Author Correction: Rad52 prevents excessive replication fork reversal and protects from nascent strand degradation. Nat. Commun. 2019, 10, 2023.
- Spies, J.; Lukas, C.; Somyajit, K.; Rask, M.-B.; Lukas, J.; Neelsen, K.J. 53BP1 nuclear bodies enforce replication timing at under-replicated DNA to limit heritable DNA damage. Nat. Cell Biol. 2019, 21, 487–497.
- Verma, P.; Dilley, R.L.; Zhang, T.; Gyparaki, M.T.; Li, Y.; Greenberg, R.A. RAD52 and SLX4 act nonepistatically to ensure telomere stability during alternative telomere lengthening. Genes Dev. 2019, 33, 221–235.
- Zhang, J.-M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968.e3.
- Kagawa, W.; Kurumizaka, H.; Ikawa, S.; Yokoyama, S.; Shibata, T. Homologous Pairing Promoted by the Human Rad52 Protein. J. Boil. Chem. 2001, 276, 35201–35208.
- Lloyd, J.A.; McGrew, D.A.; Knight, K.L. Identification of Residues Important for DNA Binding in the Full-length Human Rad52 Protein. J. Mol. Boil. 2005, 345, 239–249.
- Lloyd, J.A.; Forget, A.L.; Knight, K.L. Correlation of Biochemical Properties with the Oligomeric State of Human Rad52 Protein. J. Boil. Chem. 2002, 277, 46172–46178.
- Shen, Z.; Cloud, K.G.; Chen, D.J.; Park, M.S. Specific Interactions between the Human RAD51 and RAD52 Proteins. J. Boil. Chem. 1996, 271, 148–152.
- Ludwig, D.L.; Park, M.S.; Stigger, E.; Lee, S.-H. Physical Interaction between Human RAD52 and RPA Is Required for Homologous Recombination in Mammalian Cells. J. Boil. Chem. 1996, 271, 18996–19000.
- Altmannova, V.; Eckert-Boulet, N.; Arneric, M.; Kolesar, P.; Chaloupkova, R.; Damborsky, J.; Sung, P.; Zhao, X.; Lisby, M.; Krejci, L. Rad52 SUMOylation affects the efficiency of the DNA repair. Nucleic Acids Res. 2010, 38, 4708–4721.
- Xue, C.; Greene, E.C. New roles for RAD52 in DNA repair. Cell Res. 2018, 28, 1127–1128.
- Honda, M.; Okuno, Y.; Yoo, J.; Ha, T.; Spies, M. Tyrosine phosphorylation enhances RAD52-mediated annealing by modulating its DNA binding. EMBO J. 2011, 30, 3368–3382.
- Lim, K.; Stewart, J.; Kelly, V.; Xie, J.; Brock, K.K.; Moseley, J.; Cho, Y.-B.; Fyles, A.; Lundin, A.; Rehbinder, H.; et al. Dosimetrically Triggered Adaptive Intensity Modulated Radiation Therapy for Cervical Cancer. Int. J. Radiat. Oncol. 2014, 90, 147–154.
- Yasuda, T.; Kagawa, W.; Ogi, T.; Kato, T.A.; Suzuki, T.; Dohmae, N.; Takizawa, K.; Nakazawa, Y.; Genet, M.D.; Saotome, M.; et al. Novel function of HATs and HDACs in homologous recombination through acetylation of human RAD52 at double-strand break sites. PLoS Genet. 2018, 14, e1007277.
- Stasiak, A.Z.; Larquet, E.; Stasiak, A.; Müller, S.; Engel, A.; Van Dyck, E.; West, S.C.; Egelman, E.H. The human Rad52 protein exists as a heptameric ring. Curr. Boil. 2000, 10, 337–340.
- Kagawa, W.; Kurumizaka, H.; Ishitani, R.; Fukai, S.; Nureki, O.; Shibata, T.; Yokoyama, S. Crystal Structure of the Homologous-Pairing Domain from the Human Rad52 Recombinase in the Undecameric Form. Mol. Cell 2002, 10, 359–371.
- Singleton, M.R.; Wentzell, L.M.; Liu, Y.; West, S.C.; Wigley, D.B. Structure of the single-strand annealing domain of human RAD52 protein. Proc. Natl. Acad. Sci. USA 2002, 99, 13492–13497.
- Parsons, C.A.; Baumann, P.; Van Dyck, E.; West, S.C. Precise binding of single-stranded DNA termini by human RAD52 protein. EMBO J. 2000, 19, 4175–4181.
- Honda, M.; Wright, R.; Okuno, Y.; Rothenberg, E.; Ha, T.; Spies, M.; Grimme, J.M.; Mazin, A.V. Human Rad52 binds and wraps single-stranded DNA and mediates annealing via two hRad52-ssDNA complexes. Nucleic Acids Res. 2010, 38, 2917–2930.
- Kagawa, W.; Kagawa, A.; Saito, K.; Ikawa, S.; Shibata, T.; Kurumizaka, H.; Yokoyama, S. Identification of a Second DNA Binding Site in the Human Rad52 Protein. J. Boil. Chem. 2008, 283, 24264–24273.
- Brouwer, I.; Zhang, H.; Candelli, A.; Normanno, D.; Peterman, E.J.; Wuite, G.J.; Modesti, M. Human RAD52 Captures and Holds DNA Strands, Increases DNA Flexibility, and Prevents Melting of Duplex DNA: Implications for DNA Recombination. Cell Rep. 2017, 18, 2845–2853.
- Mazina, O.M.; Keskin, H.; Hanamshet, K.; Storici, F.; Mazin, A.V. Rad52 Inverse Strand Exchange Drives RNA-Templated DNA Double-Strand Break Repair. Mol. Cell 2017, 67, 19–29.e3.
- Zaitsev, E.N.; Kowalczykowski, S.C. A novel pairing process promoted by Escherichia coli RecA protein: Inverse DNA and RNA strand exchange. Genome Res. 2000, 14, 740–749.
- Hengel, S.R.; Malacaria, E.; Constantino, L.F.D.S.; E Bain, F.; Diaz, A.; Koch, B.G.; Yu, L.; Wu, M.; Pichierri, P.; Spies, M.A.; et al. Small-molecule inhibitors identify the RAD52-ssDNA interaction as critical for recovery from replication stress and for survival of BRCA2 deficient cells. eLife 2016, 5, 5.
- LaFargue, C.J.; Molin, G.Z.D.; Sood, A.K.; Coleman, R.L. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 2019, 20, e15–e28.
- Chun, J.; Buechelmaier, E.S.; Powell, S.N. Rad51 Paralog Complexes BCDX2 and CX3 Act at Different Stages in the BRCA1-BRCA2-Dependent Homologous Recombination Pathway. Mol. Cell. Biol. 2013, 33, 387–395.
- Whelan, D.R.; Lee, W.T.C.; Yin, Y.; Ofri, D.M.; Bermudez-Hernandez, K.; Keegan, S.; Fenyo, D.; Rothenberg, E. Spatiotemporal dynamics of homologous recombination repair at single collapsed replication forks. Nat. Commun. 2018, 9, 3882.
- Hromas, R.; Kim, H.-S.; Sidhu, G.; Williamson, E.; Jaiswal, A.; Totterdale, T.A.; Nole, J.; Lee, S.-H.; Nickoloff, J.A.; Kong, K.Y. The endonuclease EEPD1 mediates synthetic lethality in RAD52-depleted BRCA1 mutant breast cancer cells. Breast Cancer Res. 2017, 19, 122.
- Chandramouly, G.; McDevitt, S.; Sullivan, K.; Kent, T.; Luz, A.; Glickman, J.F.; Andrake, M.; Skorski, T.; Pomerantz, R.T.; Chandramouly, G. Small Molecule Disruption of RAD52 Rings as a Mechanism for Precision Medicine in BRCA Deficient Cancers. Chem. Boil. 2015, 22, 1491–1504.
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016, 44, 4189–4199.
- Sullivan, K.; Cramer-Morales, K.; McElroy, D.L.; Ostrov, D.A.; Haas, K.; Childers, W.; Hromas, R.; Skorski, T. Identification of a Small Molecule Inhibitor of RAD52 by Structure-Based Selection. PLoS ONE 2016, 11, 0147230.
- Cramer-Morales, K.; Nieborowska-Skorska, M.; Scheibner, K.; Padget, M.; Irvine, D.A.; Sliwinski, T.; Haas, K.; Lee, J.; Geng, H.; Roy, D.; et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood 2013, 122, 1293–1304.
- Li, J.; Yang, Q.; Zhang, Y.; Huang, K.; Sun, R.; Zhao, Q. Compound F779-0434 causes synthetic lethality in BRCA2-deficient cancer cells by disrupting RAD52–ssDNA association. RSC Adv. 2018, 8, 18859–18869.
- Sullivan-Reed, K.; Bolton-Gillespie, E.; Dasgupta, Y.; Langer, S.; Siciliano, M.; Nieborowska-Skorska, M.; Hanamshet, K.; Belyaeva, E.A.; Bernhardy, A.J.; Lee, J.; et al. Simultaneous Targeting of PARP1 and RAD52 Triggers Dual Synthetic Lethality in BRCA-Deficient Tumor Cells. Cell Rep. 2018, 23, 3127–3136.
- Wang, H.; Li, S.; Oaks, J.; Ren, J.; Li, L.; Wu, X. The concerted roles of FANCM and Rad52 in the protection of common fragile sites. Nat. Commun. 2018, 9, 2791.


