Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sanjeet Singh Avtaar Singh | + 4666 word(s) | 4666 | 2021-08-06 15:52:41 | | | |
| 2 | Adelaide Iervolino | Meta information modification | 4666 | 2021-09-14 11:47:37 | | | | |
| 3 | Conner Chen | Meta information modification | 4666 | 2021-09-16 03:55:22 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Avtaar Singh, S.S.; Iervolino, A. COVID-19 Pathogenesis. Encyclopedia. Available online: https://encyclopedia.pub/entry/14143 (accessed on 09 August 2026).
Avtaar Singh SS, Iervolino A. COVID-19 Pathogenesis. Encyclopedia. Available at: https://encyclopedia.pub/entry/14143. Accessed August 09, 2026.
Avtaar Singh, Sanjeet Singh, Adelaide Iervolino. "COVID-19 Pathogenesis" Encyclopedia, https://encyclopedia.pub/entry/14143 (accessed August 09, 2026).
Avtaar Singh, S.S., & Iervolino, A. (2021, September 13). COVID-19 Pathogenesis. In Encyclopedia. https://encyclopedia.pub/entry/14143
Avtaar Singh, Sanjeet Singh and Adelaide Iervolino. "COVID-19 Pathogenesis." Encyclopedia. Web. 13 September, 2021.
Copy Citation
The Coronavirus 2 (SARS-CoV-2) infection is a global pandemic that has affected millions of people worldwide. The advent of vaccines has permitted some restitution. Aside from the respiratory complications of the infection, there is also a thrombotic risk attributed to both the disease and the vaccine.
SARS-CoV-2
COVID-19
thromboembolism
ACE inhibition
pathophysiology
1. Angiotensin-Converting Enzyme (ACE) 2 Receptor: The Evolutionary Stage of Infection from Himalayan Palm Civet and Bat Coronavirus to SARS-CoV2 Infection
The gateway for SARS-CoV-2 to target cells is the angiotensin-converting enzyme (ACE) 2 receptor, which is mostly expressed by epithelial cells of the lung, heart, blood vessels, kidneys, and intestines. The ACE family of receptors includes both ACE and ACE2 which, although they both are dipeptidyl mono-carboxydipeptidases have distinct physiological functions.
2. Structure of the ACE as Ligand-Binding Receptors
SARS-CoV-2 uses common cellular transmission which is based on the binding of ligands to specific cell surface receptors. ACE2 is a G protein-coupled receptor (GPCR) and belongs to a category of receptors that play a central role in the initiation and regulation of cellular processes [1]. The GPCR constitutes the most prominent class of receptors implicated in pathological disorders of the cardiovascular, respiratory, endocrine, immune, and neural systems. Activation of GPCRs is also common in neoplastic pathologies. The function that GPCRs exert is mediated by responses to specific interactions with hormones, neurotransmitters, pathogens, metabolites, ions, fatty acids, and drugs [2][3].
GPCRs are crucial modulators of transmission between the internal and external environment of cells. GPCRs are integral membrane proteins with an extracellular N-terminal and seven transmembrane (TM) helical domains, from TM1 to TM7, connected via link regions. Evidence suggested that GPCRs have a more complex role than originally considered. The binding of GPCRs to very different types of extracellular stimuli leads to conformational changes of the TM domain with the consequent structural remodeling of the protein [4][5][6][7][8]. Inter alia, these conformation changes induce coupling with cytoplasmic proteins and subsequently the activation of enzymes that lead to the generation of a second messenger. Once the second messenger is formed it can activate a sequence of signals inside the cell [5]. This specific role of GPCRs results in increasing levels of intracellular cyclic Adenosine Monophosphate (cAMP) and represents the pivotal pathway in response to ligands, such as signaling of the renin–angiotensin system (RAS). It is important to underline that the levels of cAMP production in the cellular domain are modulated by several factors. Multidrug Resistance Proteins (MRPs) allow the efflux of cAMP from the inside of the cell to the extracellular fluid, thus maintaining homeostatic intracellular concentrations. The role of transporters activated by MRPs serves to regulate the balance of cAMP within the cell.
Lu et al. [6] reported concern about the structural conformation of the ACE/GPCR complex and its interaction with SARS-CoV by focusing on lipid rafts. The structure, activation, and signaling of the ACE/GPCR complex are strongly influenced by the bilayer domain with specific membrane-GPCR interactions [7]. It has been shown that some subsets of GPCR are preferentially isolated in distinct regions of the membrane defined as lipid rafts [8][9][10]. Cholesterol partitions preferentially into lipid rafts which contain 3 to 5-fold the amount of cholesterol found in the surrounding bilayer. Evidence has shown that lipid rafts serve as an entry site for SARS-CoV. For example, lipid rafts in Vero E6 cells were involved in the “entry” of the coronavirus of the severe acute respiratory syndrome (SARS-CoV). As has been clarified by the tests after SARS-CoV infection, the integrity of the lipid rafts was a necessary requirement to produce the pseudotyped SARS-CoV infection. If plasma membrane cholesterol depletion was induced using the relocalized MbetaCD marker on the caveolin raft the SARS-CoV, ACE2 receptor was not significantly modified. Although the surface expression of ACE2 still allowed binding to the virus, treatment with MbetaCD inhibited the infectivity of the pseudotyped SARS-CoV by 90%. The observed data concern the ectodomain of the SARS-CoV protein S (S1188HA) which can be associated with lipid rafts. The spike protein, after binding to its receptor, colocalized with the ganglioside marker GM1 residing on the raft. The study found that S1188HA binding was not affected by plasma membrane cholesterol depletion supporting the conclusion that lipid rafts serve as a gateway for SARS-CoV [11][12][13][14][15].
3. Function of ACE Receptor
The function of ACE is to split angiotensin I into angiotensin II, which binds and activates the type 1 angiotensin II receptor. This activation triggers a series of pathophysiological mechanisms that ultimately have vasoconstrictor, proinflammatory, and pro-oxidative effects. It is important to underline that among the functions of ACE2 is the hydrolytic degradation of angiotensin II to angiotensin 1–7 and angiotensin I to angiotensin 1–9. Once angiotensin 1–9 is generated, it binds to the Mas receptor, producing anti-inflammatory, antioxidant, and vasodilatory reactions. From a pathophysiological point of view, it is important to distinguish the two forms of ACE2 receptors. The first is a type 1 integral transmembrane protein with structural features representing the extracellular domain that acts as a receptor for the SARS-CoV-2 spike protein. The second is a soluble form representing circulating ACE2. To date, our knowledge is limited on the relationship that is established between SARS-CoV-2 and the two forms of the receptor. A better understanding of this relationship may more precisely define the operational adaptive or maladaptive processes that sustained COVID-19 infection [16][17].
4. ACE Receptor and Binding to Human Coronary Viruses
The knowledge we have on the interaction between the human ACE2 receptor (hACE2) and the Himalayan palm civet receptor (cACE2) with SARS-CoV derives from the usage of the receptor by the Human (hSARS-CoV) and Himalayan palm civet coronavirus (cSARS-CoV) [18]. The hSARS- CoV can bind both hACE2 and cACE2 receptors while the palm civet coronavirus has no interaction with the ACE2 receptor expressed in humans. It is known that the adaptation of c SARS-CoV to humans was determined by two-point mutations, recognized as K479N and S487T, in the binding domain of the SARS-CoV spike protein (SARS-CoV-S) [17].
The mutations that have recently characterized SARS-CoV-2 led to more aggressive variants of the virus and the concept of adaptive mutations (as noted by Wu et al. [19]) with strengthened receptor binding and tropism (RBT). The authors demonstrated that adaptive mutations of RBT led to the identification of genetic mutations of the virus that enhanced interaction with human or palm civet ACE2. The genetic adaptation processes that took place between hSARS-CoV and cSARS-CoV could also be recorded in SARS-CoV-like viruses that have been isolated in bats [19]. A previous study found that the pathways in which bat coronaviruses infected host cells did not occur through the interaction of the ACE2 receptor with expressed SARS-CoVS and remained a mystery. However, the important finding remains that the substitution of the amino acid sequence found between residues 323 and 505 of the corresponding sequence of the SARS-CoV-S/RBT is sufficient to allow the activation of the human ACE2 receptor [19].
Coronaviruses can enter target cells effortlessly due to their ability to exploit many cell surface molecules such as proteins and carbohydrates. Lectins play a fundamental role in this process. For example, host calcium-dependent (type C) lectins have been recognized to play a central role in SARS-CoV-2 infection. Evidence suggests a specific intercellular role exerted by non-integrin 3-grabbing adhesion molecule (DC-SIGN) of dendritic cells. This is a type C lectin expressed on macrophages and dendritic cells that functions to recognize the high-mannose glycosylation patterns commonly found on viral and bacterial pathogens. Coronavirus protein S is highly glycosylated, thus, providing the virus with the opportunity to interact with host lectins such as Dendritic Cell (DC)/Liver/lymph node-specific intercellular adhesion molecule-3-grabbing integrin (L-SIGN). L-SIGN, which is expressed on liver and lung endothelial cells and has been reported as an alternative receptor for SARS-CoV and bat coronavirus type HCoV-229E [20][21].
The first demonstration of the possibility that SARS-CoV-2 interacts with the human ACE2 receptor is reported in the landmark study from the University of North Carolina at Chapel Hill [22][23]. The authors reported the substantially high risk of SARS-like bat coronavirus disease named SHC014-CoV circulating in Chinese horseshoe bat populations. This type of coronavirus has a high binding affinity with the ACE2 receptor [24][25]
The new SARS-CoV-2 virus expressed the bat coronavirus SHC014 spike in mouse-adapted SARS-CoV backbones.
Menachery et al. created a chimeric virus starting with the RsSHC014-CoV sequence that was isolated from Chinese horseshoe bats [25]. The chimeric virus encoded a different, zoonotic CoV spike protein in the context of the SARS-CoV mouse-adapted backbone. This new SARS-CoV-2 virus expressed the bat coronavirus SHC014 spike. Through the hybrid virus, the authors were able to evaluate the ability of the new spike protein to cause disease independently of other necessary adaptive mutations in its natural backbone [23].
The evidence showed at least two very interesting findings. The first was that group 2b viruses encoding the SHC014 peak in a wild-type backbone could efficiently use more orthologs of the human angiotensin II converting enzyme (ACE2) receptor than the unmodified SARS virus. The second was that group 2b viruses could replicate efficiently in primary human airway cells and that it was also possible to obtain in vitro viral titers equivalent to the epidemic strains of SARS-CoV. Once these results were translated in vivo, replication of the chimeric virus in the mouse lung demonstrated considerable pathogenesis. This led to the trials of immunotherapeutic and prophylactic modalities to cope with the SARS-CoV infection which had poor outcomes. In fact, both monoclonal antibodies and the vaccine approach failed to neutralize and protect against CoV infection using the new SARS-CoVS. Based on these results, the authors synthetically re-derived an infectious full-length recombinant SHC014 virus and demonstrated robust viral replication both in vitro and in vivo settings. This landmark report suggested 6 years ago that there was a potential risk of SARS-CoV re-emergence from viruses circulating in bat populations.
Recently the same group coordinated by Ralph Baric [26] studied the critical determinants of the ACE2 receptor that support SARS-CoV-2-ACE2 interactions during infection and replication of the preemergent 2B coronavirus (WIV). The Authors identified the key changes that lead to infection by creating a humanized murine ACE2 receptor (hmACE2) and provided evidence for the potential pan-virus capabilities of this chimeric receptor.
SARS-CoV-2 cannot infect mice due to incompatibility between its receptor-binding domain (RBD) and the murine ACE2 (mACE2) receptor. Since the mouse models of human ACE2 (hACE2) and viruses adapted to mice have shown limitations, the researchers developed another model that would allow evaluation of the pathogenetic phenomena that occur in human SARS-CoV-2 infection. For example, hACE2 transgenic mice are susceptible to unadapted SARS-CoV-2 viruses, but the pathogenesis observed in these mice showed that virus-induced encephalitis and multi-organ infection were not comparable to that observed in humans. Thus, Adams et al., to map the SARS-CoV-2 RBD and mACE2 interaction network, created a panel of mACE2 receptors, which have increasing levels of humanizing mutations. The study used predictive structural models that allowed identification of the minimal changes needed to restore replication [26].
The ACE2 receptor has structurally critical sites whose integrity determines its activity. The investigators worked at the level of three hot spots that determine ACE2 interaction: position K353 interconnects with SARS-CoV-2 binding residues G496, N501, and Y505, position K31 which forms a salt bridge with ACE2 residue K353 and links with SARS-CoV-2 Q493 and Y489, and position M82 which interconnects with RBD residues F486, N487, and Y489. These aforementioned interface hotspots are the critical molecular sites for the interaction between SARS-CoV2 and the receptor leading to virus entry. The authors demonstrated that divergent residue modifications in these hot spots significantly reduce the binding between humanized murine ACE2 (hmACE2) [24] and SARS-CoV-2 RBD. They recorded that five amino acid changes (N30D, N31K, S82M, F83Y, and H353K) in the SARS-CoV-2 RBD-ACE2 interaction hot spots lead to the modulation of infection and can re-establish infection in the hmACE2 models [26].
This study is crucial for the following reasons. The first is related to the fact that mouse models are essential for understanding the pathogenesis of coronaviruses and are a key resource for the preclinical development of vaccines and antiviral therapies. The second is that a detailed analysis of this study will allow the development of model systems to screen for emerging coronaviruses and to develop new treatments to combat infections [26].
5. The Role of ACE2 in COVID-19 Pathogenesis
The ACE2 receptor has been implicated in the pathogenesis of COVID-19, especially with regards to its potential effects on the most vulnerable patients presenting with cardiovascular co-morbidities. COVID-19 does not have the same impact on all members of the population. An exponential increase in the severity of the disease as well as mortality, due to devastating thromboembolic complications, occurs in patients over the sixth decade of life with comorbidities such as cardiovascular disease and diabetes.
The angiotensin-converting-enzyme 2 receptors (ACE2) serve as the attachment site of the SARS-CoV-2 spike protein to enter the lung epithelial cells [27]. Upregulation with increased ACE2 expression has been demonstrated in patients with cardiovascular disease and diabetes treated with angiotensin-converting enzyme (ACEI) inhibitors and angiotensin receptor blockers (ARBs). However, whether treatment with these agents can lead to greater COVID-19 severity has not been fully clarified.
Discussions related to the use of ACEI/ARBs have surfaced regarding the need to continue therapy in patients taking these drugs. The current recommendations are to discontinue the administration of these drugs, despite diverging opinions, which were not universally endorsed by experts due to the lack of strong evidence [28]. ACE2 not only plays a role in the pathogenesis of COVID-19 but also as a component of renin–angiotensin system signaling (RAS) localized throughout the body. Although the evidence has conclusively revealed that ACE2 receptors allow SARS-CoV-2 to enter cells, ACE2 plays a central anti-inflammatory role in RAS signaling by converting angiotensin II, responsible for the inflammatory process, into angiotensin 1–7, which leads to its anti-inflammatory effects [29]. A study performed on rodent lungs [30] showed that the reduced expression of ACE2 leads to a sequence of major proinflammatory processes, that are exacerbated by age, and result in dysregulation of RAS signaling throughout the body [31]. It is important to note that this typical inflammatory profile, even in accentuated forms, supports pathophysiological processes that represent the main feature of hypertension and diabetes, as well as being very widespread in old age [27]. The upregulation of the ACE2 receptor in subjects with diabetes and hypertension treated with ACI/ARBs must be seen as a restorative substrate that has a physiological function. The process that unfolds during SARS-CoV2 infection sees ACE2 receptors as a gateway for the virus to enter cells, while the reduction of ACE2 protective features in older people and those with CVD can potentially predispose them to more severe forms of the disease. The ACE2 receptor facilitates SARS-CoV2 infection while the fundamental anti-inflammatory function, linked to RAS signaling, is reduced because it is compromised in patients who develop COVID-19. In fact, data provided by the first SARS epidemic in 2003 demonstrated the double role of the ACE2 receptor, thus delineating the factors predisposing to the occurrence of the disease and its severity [32][33].
In SARS-CoV2 infection it is plausible that the higher expression of ACE2 leads to a greater predisposition to experience the disease. Epidemiological data from the South Korean population, where genetic testing has been widely used in individuals, reported higher numbers of infected among young adults [32] and those with increased ACE2 levels. In this regard, an Italian study, examining the severity of COVID-19 disease in the elderly population with CVD, hypothesized that a reduction in ACE2 levels due to aging and CVD coupled to the upregulation of the proinflammatory angiotensin II pathway are factors that likely predispose older individuals to severe forms of COVID-19. Therefore, younger people are more susceptible to viral infection, but older people are more likely to have severe disease manifestations [33].
SARS-CoV2 uses the ACE2 receptor in carrying out its infectious manifestation, thereby leading to a reduced expression of ACE2 on the cell surface and an upregulation of angiotensin II signaling in the lungs which results in the development of acute damage [29]. The consequence of these morphofunctional and biochemical changes can predispose elderly individuals with CVD, who have reduced levels of ACE2 compared to young people, to exaggerated inflammation and further reduction of ACE2 expression in the context of COVID-19. In these cases, the disease manifests itself with greater severity [34]. Observations suggest that older individuals, especially those with hypertension and diabetes, have reduced ACE2 expression and upregulation of proinflammatory angiotensin II signaling. Therefore, the morphofunctional and biochemical changes can be corrected by the increase in ACE2 levels induced by ACEI/ARB treatment [34].
First, it is possible to hypothesize that in COVID-19 disease, the binding of SARS-CoV-2 to ACE2 receptors acutely exacerbates this proinflammatory background, predisposing these subpopulations to greater severity and mortality of COVID-19 disease. Second, considering this hypothesis credible, a protective role of the antagonistic action of angiotensin II against acute lung injury associated with sepsis could be effective. This supports the use of continuous therapy with ACEI/ARB [35][36][37] (Figure 1).
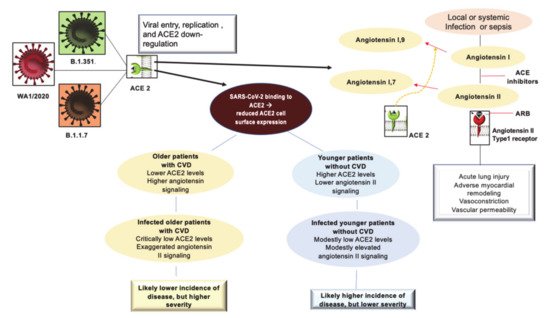
Figure 1. Depicts the interaction of SARS-CoV2 with the ACE2 receptor and the inflammatory profile pattern before and after Coronavirus 2019 (COVID-19) infection in patients with or without CVD. The initial entry of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) into cells is shown with involvement mainly of type II pneumocytes. SARS-CoV-2 binds to its functional receptor, the angiotensin-converting enzyme 2 (ACE2). After endocytosis of the viral complex, surface ACE2 is further down-regulated, resulting in unobstructed accumulation of angiotensin II. Local activation of the renin–angiotensin–aldosterone system may mediate lung injury responses to viral insults. The elderly and the young may present with different pathophysiological profiles. The simplified scheme of the pre-infection inflammatory profile among predisposed older individuals compared to their younger counterparts is illustrated. Abbreviations: ACE2, angiotensin-converting enzyme 2; ARB, angiotensin-receptor blocker; CVD, cardiovascular disease; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Third, the aforementioned biomechanical modifications of the receptor, plausible with aging, should be investigated. Therefore, experiments on the functioning, the regulatory mechanisms of RAS, and the biomechanics of the receptors involved in these functions should be implemented. Specifically, the biophysical mechanisms underlying the associated remodeling of the lipid membrane remain to be clarified. They may be useful in the prevention of fatal lung complications caused by genetic variants of the Wuhan virus [17][18][19][23][25][26][38].
6. ACE Inhibitors and Angiotensin Type II Blockers Role in COVID-19 Severity
Tetlow et al. [39] did not identify any associations between ACE-I/ARB use and AKI, macrovascular thrombi, or mortality. Other studies [40][41] also supported the continuation of these drugs during hospitalization from COVID-19. Among those hospitalized, a large percentage are likely being administered either ACE inhibitors or Angiotensin II blockers, since epidemiological data reveal that cardiomyopathies, diabetes mellitus, and hypertension are the most frequent comorbidities found among those patients [42].
Although the upregulation of ACE2 expression, which can be altered by drug administration, has not been defined, it has nevertheless been associated with disease severity. Several preceding studies have demonstrated that the risk of developing COVID-19 after the administration of ACEi and ARBs increased significantly. This could be an indirect effect of overproduction of the circulating ACE2 transcripts in the cells [43][44]. As an example, Enalapril, which is a frequently used ACEi, was reported to increase ACE2 expression in the kidney [45].
Concerning possible therapeutical targets, ACE2 blockers have been developed, such as the small synthetic inhibitor N-(2-aminoethyl)-1aziridine-ethanamine (NAAE) [46]. It is able to bind ACE2 in its closed conformation so that molecular interaction between the viral particle and the receptor cannot be possible and fusion does not happen. Thus, NAAE could exert dual inhibitory effects: one on ACE2 catalytic activity and another on SARS binding [47]. Despite this, current research drives opinions towards a cautionary use of this agent.
7. Pathophysiology of Arterial and Venous Thrombosis
To date, the complete pathophysiology profile of arterial and venous thrombosis during COVID-19 disease has not yet been fully clarified. The literature reports prothrombotic abnormalities in patients with COVID-19. In a Chinese study [48] performed in the first phase of the SARS-CoV2 epidemic, 19 patients with COVID-19, who presented with critical clinical conditions, had elevated levels of markers of hypercoagulability such as D-dimer found in 100%, fibrinogen in 74%, and factor VIII in 100%. The dysregulation of the coagulation process included the presence of antiphospholipid antibodies in 53% of the population studied. Reduced levels of protein C, protein S, and antithrombin were noted in all patients. Complications such as stroke, arterial ischaemia, and VTE accompanied the coagulation disorder.
Zaid et al. studied 115 patients with COVID-19 disease reporting that SARS-CoV2 directly interferes with platelets. Viral RNA and high platelet-associated cytokine levels were found in the platelets of all study participants. These abnormalities were not related to the severity of the disease because in 71 infected individuals the disease manifested in a non-serious manner while for 44 patients’ hospitalization was required for critical clinical conditions. Specific tests performed on platelets showed aggregation at lower than expected thrombin concentrations [49].
Nicolai et al. examined the autopsy findings of 38 individuals who died with COVID-19 which showed that histopathological changes in coagulation were marked in the vessel microcirculation. The abnormalities recorded were microvascular thrombotic formations, and neutrophil extracellular traps characterized by networks of extracellular neutrophil-derived DNA and polymorphonuclear neutrophil (PMN)-platelet aggregates [50]. The authors compared the peripheral blood of patients with COVID-19 with that of healthy patients. In vitro responses on peripheral blood samples from the three infected patients exhibited excessive platelet and neutrophil activation, as assessed by degranulation and integrin IIb-IIIa activation and immunofluorescence, compared to healthy control patients’ samples.
7.1. The Inflammatory Response during SARS-CoV2 Infection and Thrombotic Complication
Histopathology of SARS infection Cov2 is distinguished from that caused by other viruses with tropism for the respiratory tract. SARS-CoV2 leads to direct damage of endothelial cells characterized by dense perivascular infiltration of T lymphocytes combined with aberrant activation of macrophages. The excessive and uncontrolled inflammatory response, endothelial cell apoptosis, thrombotic microangiopathy, and angiogenesis are other distinctive histopathological features that denote the aggressiveness of SARS-CoV2, which may be responsible for clinically severe forms of COVID-19 thus conferring disease characteristics not comparable to any other viral respiratory disorder [51]. One significant finding that emerges in the evaluation of the pathophysiology of thromboembolism in COVID-19 versus non-COVID-19 disorders is the possibility that the coagulation alterations are mediated more by platelet-dependent activation and intrinsically related to viral-mediated endothelial inflammation. As noted a distinguishing feature of thrombosis during SARS-CoV2 infection is the exacerbated hypercoagulability associated with increased concentrations of coagulation factors, acquired antiphospholipid antibodies, and reduced concentrations of endogenous anticoagulant proteins [48].
Patients with COVID-19 who develop more severe systemic inflammation and more critical respiratory dysfunction have a higher prevalence of thrombotic complications. Lodigiani et al. reported 388 patients hospitalized with COVID-19 including 16% with serious clinical conditions. Despite the use of low molecular weight heparin (LMWH) for thromboprophylaxis in all patients in the ICU and 75% of those not in the ICU, symptomatic VTE occurred in 4.4% of patients, ischemic stroke in 2.5%, and MI in 1.1% [52].
Given the knowledge we have, there is still no clarity on the extent to which SARS-CoV-2 increases the risk of thromboembolism. A study performed in the United Kingdom compared 1877 patients discharged from hospital after COVID-19 disease and 18,159 hospitalized for a non-COVID-19 disease reported no difference in hospital-associated VTE rates (4.8/1000 vs. 3.1/1000; odds ratio, 1.6 [95% CI, 0.77–3.1]; p = 0.20) [53]. One point to clarify is whether the high rate of VTE is specific to patients who develop COVID-19 or if VTE is mainly occurring in patients as a complication associated with severe critical disease [53]. These results are in line with a recent meta-analysis that included 41,768 patients in whom VTE was assessed in COVID-19 versus non-COVID-19 cohorts. The authors did not record a significant statistical difference for overall risk of VTE (RR 1.18; 95%CI 0.79–1.77; p = 0.42; I2 = 54%), pulmonary embolism (RR 1.25; 95%CI 0.77–2.03; p = 0.36; I2 = 52%) and deep venous thrombosis (RR 0.92; 95%CI 0.52–1.65; p = 0.78; I2 = 0%). A difference was reported after analyzing the subgroups of patients who were admitted to the intensive care unit (ICU). Critically ill patients had an increased risk of VTE in the COVID-19 cohort compared to non-COVID-19 patients admitted to the ICU (RR 3.10; 95% CI 1.54–6, 23), which was not observed in cohorts of non-ICU patients (RR 0.95; 95% CI 0.81–1.11) (P interaction = 0.001) [54].
7.2. Management of Thrombosis in COVID-19 Patients
There are no international guidelines that direct the prevention and treatment of thrombotic complications in COVID-19 patients. Both published and ongoing studies testing interventions to prevent thrombosis complications in COVID-19 are based on the evidence reported in current clinical guidelines about VTE prophylaxis in acute COVID-19 infections. Therefore, pending the results to be provided by the completion of ongoing trials, guidelines for the treatment of thrombotic complications in patients with COVID-19 disease are derived from medical recommendations in the coagulation disorder populations (Figure 2). However, the crucial point that remains to be clarified is whether these guidelines are also optimal for the treatment of thrombosis due to COVID-19 [55][56][57].
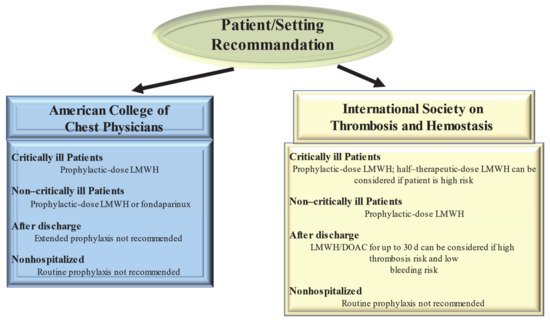
Figure 2. Current Guideline Recommendations for Venous Thromboembolism Prevention in patients With Coronavirus Disease 2019. Abbreviations: DOAC, direct oral anticoagulant; LMWH, low-molecular-weight heparin.
Guidelines from the American College of Chest Physicians (ACCP) suggest (in the absence of contraindications) prophylaxis with LMWH or fondaparinux rather than unfractionated heparin or direct oral anticoagulants (DOACs) for all hospitalized COVID-19 patients [55] Clearly the optimum choice of the drug to be taken is constrained by the incomplete knowledge of the possible interference of CoV 2 SARS with the medicament. So, the 40 mg dose of LMWH for injection once a day and the 2.5 mg dose of fondaparinux are preferred over the administration of unfractionated heparin injected subcutaneously 2–3 times a day thus limiting the caregiver’s contact with infected patients. In addition, these drugs are preferred over DOACs because of drug–drug interactions with antiviral agents. Both are substrates of the P-glycoprotein and/or cytochrome P450-based metabolic pathways. Thus, concomitant administration of DOACs and antiviral drugs has the potential to sharply increase DOAC anticoagulant plasma levels, thus increasing hemorrhagic risk.
Given the high incidence of VTE, the proposed therapeutic dose to be used for standard thromboprophylaxis in critically ill patients with COVID-19 was either double or single-dose administration of LMWH. The ACCP guidelines suggest the standard dose LWMH in the absence of new clinical trial data [56]. Guideline-Directed Medical Therapy (GDMT), which was established by the International Society on Thrombosis and Hemostasis (ISTH), suggested that half-therapeutic-dose LMWH (1 mg/kg daily) can be considered for prophylaxis in high-risk patients with COVID-19. A 50% higher dose can be considered in patients with severe obesity (BMI ≥ 40 kg/m2). However, it remains to be clarified which is the best dosage for optimal prophylactic therapy [57]. The results of ongoing randomized controlled trials (REMAP-CAP, ACTIV-4, and ATTACC), comparing therapeutic-intensity anticoagulation with prophylactic-intensity anticoagulation for patients with COVID-19-related critical illness, are awaited to establish optimal antithrombotic prophylactic therapy [58]. Considering the pathophysiology of thromboembolism in COVID-19 is characterized by platelet hyperreactivity, another point under discussion with RCTs initiated is the evaluation of administering an antiplatelet agent for therapeutic prophylaxis.
High-risk patients hospitalized for COVID-19 have a high possibility of developing VTE that persists after discharge [57]. However, for the latter, no specific recommendations for post-discharge thromboprophylaxis have been established by the ACCP [56]. In contrast, the ISTH recommendations for post-discharge thromboprophylaxis suggest the use of LMWH or a DOAC for all high-risk hospitalized patients with COVID-19 who have a low risk of bleeding. Patients with COVID-19 considered to be at high risk include those with age ≥ 65 years, presence of critical illness, cancer, previous VTE, thrombophilia, severe immobility, and elevated D-dimer (>2 times the upper limit of normal). ISTH recommendations suggest a duration of 14 to 30 days for post-discharge thromboprophylaxis, although the ideal administration period remains to be clarified [57].
For patients with COVID-19 disease, no diagnostic protocols have been established for thromboembolic complications, such as pulmonary embolism and MI, so the methods to be used should be those validated for patients without COVID-19. Given the lack of evidence to support the benefit, routine ultrasound checks for VTE surveillance are not recommended. For patients with COVID-19 diagnosed with arterial or venous thrombosis, we recommend treatment according to current established guidelines. These recognize the benefits of LMWH administration in hospitalized patients. In the outpatient setting, DOAC administration is recommended [50]. There are currently no recommendations issued by ISTH and ACCP to support measuring the D-dimer to screen for VTE or to establish the intensity of prophylaxis or treatment [56][57].
References
- Wichmann, D.; Sperhake, J.-P.; Lütgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, F.; Mushumba, H.; Kniep, I.; Schröder, A.S.; et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19. Ann. Intern. Med. 2020, 173, 268–277.
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847.
- Cui, S.; Chen, S.; Li, X.; Liu, S.; Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 1421–1424.
- Allard, J.F.; Dushek, O.; Coombs, D.; van der Merwe, P. Mechanical Modulation of Receptor-Ligand Interactions at Cell-Cell Interfaces. Biophys. J. 2012, 102, 1265–1273.
- Mary, S.; Fehrentz, J.; Damian, M.; Vedié, P.; Martinez, J.; Marie, K.; Baneres, J. How ligands and signaling pro-teins affect G-protein-coupled receptors’ conformational landscape. Biochem. Soc. Trans. 2013, 41, 144–147.
- Unal, H.; Karnik, S.S. Domain coupling in GPCRs: The engine for induced conformational changes. Trends Pharmacol. Sci. 2012, 33, 79–88.
- Kobilka, B.K.; Deupi, X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol. Sci. 2007, 28, 397–406.
- Lefkowitz, R.J. Seven transmembrane receptors: Something old, something new. Acta Physiol. 2007, 190, 9–19.
- Lu, Y.; Liu, D.X.; Tam, J.P. Lipid rafts are involved in SARS-CoV entry into vero E6 cells. Biochem. Biophys. Res. Commun. 2008, 369, 344–349.
- Oates, J.; Watts, A. Uncovering the intimate relationship between lipids, cholesterol and GPCR activation. Curr. Opin. Struct. Biol. 2011, 21, 802–807.
- Shukla, A. (Ed.) G Protein-Coupled Receptors. Signaling, Trafficking and Regulation, 1st ed.; Methods in Cell Biology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 132.
- Villar, V.; Cuevas, S.; Zheng, X.; Jose, P. Chapter 1: Localization and signaling of GPCRs in lipid rafts. Methods Cell Biol. 2016, 136, 3–23.
- Watkins, E.; Miller, C.; Majewski, J.; Kuhl, T. Membrane texture induced by specific protein binding and receptor clustering: Active roles for lipids in cellular function. Proc. Natl. Acad. Sci. USA 2011, 108, 6975–6980.
- Lu, Y.; Liu, D.X.; Tam, J.P. Lipid rafts play an important role in the early stage of severe acute respiratory syn-drome-coronavirus life cycle. Microbes Infect. 2007, 9, 96–102.
- Lingwood, D.; Simons, K. Lipid Rafts As a Membrane-Organizing Principle. Science 2009, 327, 46–50.
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454.
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.-K.; Huang, I.-C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643.
- Song, H.-D.; Tu, C.-C.; Zhang, G.-W.; Wang, S.-Y.; Zheng, K.; Lei, L.-C.; Chen, Q.-X.; Gao, Y.-W.; Zhou, H.-Q.; Xiang, H.; et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl. Acad. Sci. USA 2005, 102, 2430–2435.
- Wu, K.; Peng, G.; Wilken, M.; Geraghty, R.J.; Li, F. Mechanisms of host receptor adaptation by severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2012, 287, 8904–8911.
- Menachery, V.D.; Yount, B.L., Jr.; Debbink, K.; Agnihothram, S.; Gralinski, L.E.; Plante, J.A.; Graham, R.L.; Scobey, T.; Ge, X.Y.; Donaldson, E.F.; et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2015, 21, 1508–1513.
- Menachery, V.D.; Yount, B.L.; Debbink, K.; Agnihothram, S.; Gralinski, L.E.; Plante, J.A.; Graham, R.L.; Scobey, T.; Ge, X.-Y.; Donaldson, E.F.; et al. Author Correction: A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2020, 26, 1146.
- Ren, W.; Qu, X.; Li, W.; Han, Z.; Yu, M.; Zhou, P.; Zhang, S.Y.; Wang, L.F.; Deng, H.; Shi, Z. Difference in receptor usage be-tween severe acute respiratory syndrome (SARS) coronavirus and SARS-like coronavirus of bat origin. J. Virol. 2008, 82, 1899–1907.
- Ge, X.-Y.; Li, J.; Yang, X.-L.; Chmura, A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538.
- Adams, L.E.; Dinnon, K.H., 3rd.; Hou, Y.J.; Sheahan, T.P.; Heise, M.T.; Baric, R.S. Critical ACE2 Determinants of SARS-CoV-2 and Group 2B Coronavirus Infection and Replication. mBio 2021, 12, e03149-20.
- Jeffers, S.A.; Tusell, S.M.; Gillim-Ross, L.; Hemmila, E.M.; Achenbach, J.E.; Babcock, G.J.; Thomas, W.D.; Thackray, L.B.; Young, M.D.; Mason, R.J.; et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 15748–15753.
- Jeffers, S.A.; Hemmila, E.M.; Holmes, K.V. Human Coronavirus 229E can Use CD209L (L-Sign) to Enter Cells. Adv. Exp. Med. Biol. 2006, 581, 265–269.
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879.
- Bozkurt, B.; Kovacs, R.; Harrington, B. HFSA/ACC/AHA Statement Addresses Concerns Re: Using RAAS Antagonists in COVID-19. Available online: https://professional.heart.org/professional/ScienceNews/UCM_505836_HFSAAC-CA-HA-statement-addresses-concerns-reusing-RAAS-antagonists-in-COVID-19.jsp?utm_campaign=sciencenews19-20&utm_source=science-news&utm_medium=email&utm_content=phd03-17-20 (accessed on 22 March 2020).
- Imai, Y.; Kuba, K.; Penninger, J.M. The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp. Physiol. 2008, 93, 543–548.
- Xudong, X.; Junzhu, C.; Xingxiang, W.; Furong, Z.; Yanrong, L. Age- and gender-related difference of ACE2 expression in rat lung. Life Sci. 2006, 78, 2166–2171.
- Lakatta, E.G. The reality of getting old. Nat. Rev. Cardiol. 2018, 15, 499–500.
- Liang, W.; Zhu, Z.; Guo, J.; Liu, Z.; He, X.; Zhou, W.; Chin, D.P.; Schuchat, A.; Beijing Joint SARS Expert Group. Severe Acute Respiratory Syndrome, Beijing, 2003. Emerg. Infect. Dis. 2004, 10, 25–31.
- Backhaus, A. Coronavirus: Why It’s So Deadly in Italy. Available online: https://www.researchgate.net/publication/342014166_Coronavirus_Why_it’s_so_deadly_in_Italy (accessed on 22 March 2020).
- Rodrigues Prestes, T.R.; Rocha, N.P.; Miranda, A.S.; Teixeira, A.L.; Simoes-E-Silva, A.C. The anti-inflammatory potential of ACE2/angiotensin-(1-7)/Mas receptor axis: Evidence from basic and clinical research. Curr. Drug Targets 2017, 18, 1301–1313.
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116.
- Imai, Y.; Kuba, K.; Penninger, J.M. Angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Cell. Mol. Life Sci. 2007, 64, 2006–2012.
- Zhang, X.; Li, S.; Niu, S. ACE2 and COVID-19 and the resulting ARDS. Postgrad. Med. J. 2020, 96, 403–407.
- Fallahi-Sichani, M.; Linderman, J. Lipid raft-mediated regulation of G-protein coupled receptor signaling by ligands which influence receptor dimerization: Acomputational study. PLoS ONE 2009, 4, e6604.
- Tetlow, S.; Segiet-Swiecicka, A.; O’Sullivan, R.; O’Halloran, S.; Kalb, K.; Brathwaite-Shirley, C.; Alger, L.; Ankuli, A.; Baig, M.S.; Catmur, F.; et al. ACE inhibitors, angiotensin receptor blockers and endothelial injury in COVID-19. J. Intern. Med. 2020, 289, 688–699.
- Cannata, F.; Chiarito, M.; Reimers, B.; Azzolini, E.; Ferrante, G.; My, I.; Viggiani, G.; Panico, C.; Regazzoli, D.; Ciccarelli, M.; et al. Continuation versus discontinuation of ACE inhibitors or angiotensin II receptor blockers in COVID-19: Effects on blood pressure control and mortality. Eur. Heart J. Cardiovasc. Pharmacother. 2020, 6, 412–414.
- Derington, C.G.; Cohen, J.B.; Mohanty, A.F.; Greene, T.H.; Cook, J.; Ying, J.; Wei, G.; Herrick, J.S.; Stevens, V.W.; Jones, B.E.; et al. Angiotensin II receptor blocker or angiotensin-converting enzyme inhibitor use and COVID-19-related outcomes among US Veterans. PLoS ONE 2021, 16, e0248080.
- Parit, R.; Jayavel, S. Association of ACE inhibitors and angiotensin type II blockers with ACE2 overexpression in COVID-19 comorbidities: A pathway-based analytical study. Eur. J. Pharmacol. 2021, 896, 173899.
- Fang, L.; Karakiulakis, G.; Roth, M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir. Med. 2020, 8, e21.
- Watkins, J. Preventing a covid-19 pandemic. BMJ 2020, 368, m810.
- Abuohashish, H.M.; Ahmed, M.M.; Sabry, D.; Khattab, M.M.; Al-Rejaie, S.S. ACE-2/Ang1-7/Mas cascade mediates ACE inhibitor, captopril, protective effects in estrogen-deficient osteoporotic rats. Biomed. Pharmacother. 2017, 92, 58–68.
- Saponaro, F.; Rutigliano, G.; Sestito, S.; Bandini, L.; Storti, B.; Bizzarri, R.; Zucchi, R. ACE2 in the Era of SARS-CoV-2: Controversies and Novel Perspectives. Front. Mol. Biosci. 2020, 7, 588618.
- Adedeji, A.; Sarafianos, S.G. Antiviral drugs specific for coronaviruses in preclinical development. Curr. Opin. Virol. 2014, 8, 45–53.
- Zhang, Y.; Cao, W.; Jiang, W.; Xiao, M.; Li, Y.; Tang, N.; Liu, Z.; Yan, X.; Zhao, Y.; Li, T.; et al. Profile of natural anticoagulant, coagulant factor and anti-phospholipid antibody in critically ill COVID-19 patients. J. Thromb. Thrombolysis 2020, 50, 580–586.
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; et al. Platelets can associate with SARS-Cov-2 RNA and are hyperactivated in COVID-19. Circ Res. 2020, 127, 1404–1418.
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated With Respiratory Failure and Coagulopathy. Circulation 2020, 142, 1176–1189.
- Ackermann, M.; Verleden, S.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128.
- Girardi, F.M.; Barra, M.B.; Zettler, C.G. Papillary thyroid carcinoma: Does the association with Hashimoto’s thyroiditis affect the clinicopathological characteristics of the disease? Braz. J. Otorhinolaryngol. 2015, 81, 283–287.
- Roberts, L.N.; Whyte, M.B.; Georgiou, L.; Giron, G.; Czuprynska, J.; Rea, C.; Vadher, B.; Patel, R.K.; Gee, E.; Arya, R. Postdischarge venous thromboembolism following hospital admission with COVID-19. Blood 2020, 136, 1347–1350.
- Mai, V.; Tan, B.K.; Mainbourg, S.; Potus, F.; Cucherat, M.; Lega, J.-C.; Provencher, S. Venous thromboembolism in COVID-19 compared to non-COVID-19 cohorts: A systematic review with meta-analysis. Vasc. Pharmacol. 2021, 139, 106882.
- Patell, R.; Bogue, T.; Koshy, A.; Bindal, P.; Merrill, M.; Aird, W.C.; Bauer, K.A.; Zwicker, J.I. Postdischarge thrombosis and hemorrhage in patients with COVID-19. Blood 2020, 136, 1342–1346.
- Moores, L.K.; Tritschler, T.; Brosnahan, S.; Carrier, M.; Collen, J.F.; Doerschug, K.; Holley, A.B.; Jimenez, D.; LeGal, G.; Rali, P.; et al. Prevention, diagnosis and treatment of venous thromboembolism in patients with COVID-19: CHEST Guideline and Expert Panel Report. Chest 2020, 158, 1143–1163.
- Spyropoulos, A.C.; Levy, J.H.; Ageno, W.; Connors, J.M.; Hunt, B.J.; Iba, T.; Levi, M.; Samama, C.M.; Thachil, J.; Giannis, D.; et al. Scientific and Standardization Committee communication: Clinical guidance on the diagnosis, prevention, and treatment of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1859–1865.
- Cuker, A.; Tseng, E.K.; Nieuwlaat, R.; Angchaisuksiri, P.; Blair, C.; Dane, K.; Davila, J.; DeSancho, M.T.; Diuguid, D.; Griffin, D.O.; et al. American Society of Hematology 2021 guidelines on the use of anticoagulation for thromboprophylaxis in patients with COVID-19. Blood Adv. 2021, 5, 872–888.
More
Information
Subjects:
Peripheral Vascular Disease
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
3 times
(View History)
Update Date:
16 Sep 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No