 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | 國城 盧 | + 3831 word(s) | 3831 | 2021-09-13 04:41:10 | | | |
| 2 | Peter Tang | Meta information modification | 3831 | 2021-09-14 05:03:06 | | |
Video Upload Options
COVID-19 symptoms are highly heterogeneous, and the patient may be asymptomatic or may present with mild to severe or fatal symptoms. Factors, such as age, sex, and comorbidities, are key determinants of illness severity and progression. Aging is accompanied by multiple deficiencies in interferon production by dendritic cells or macrophages in response to viral infections, resulting in dysregulation of inflammatory immune responses and excess oxidative stress. Age-related dysregulation of immune function may cause a more obvious pathophysiological response to SARS-CoV-2 infection in elderly patients and may accelerate the risk of biological aging, even after recovery. Resveratrol is a potent antioxidant with antiviral activity.
1. Introduction
2. Hyperinflammation in COVID-19
3. SARS-CoV-2 Infection and Oxidative Stress
4. Innate Immune Response to COVID-19
4.1. Innate Immune Response
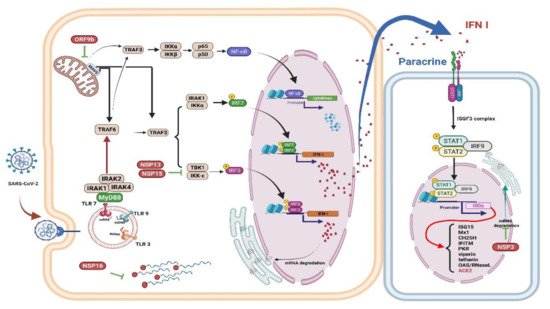
4.2. Interferons (IFNs), Proinflammatory Cytokines, and Oxidative Stress
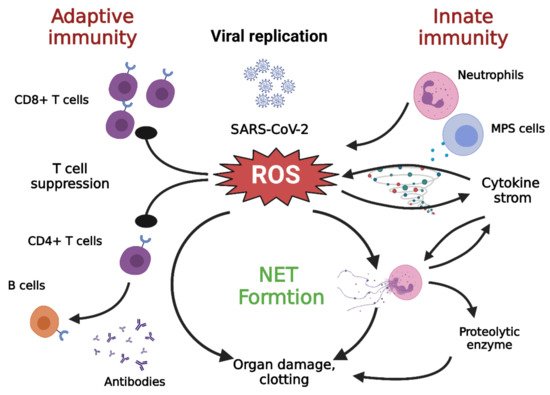
4.3. Neutrophils and NETs
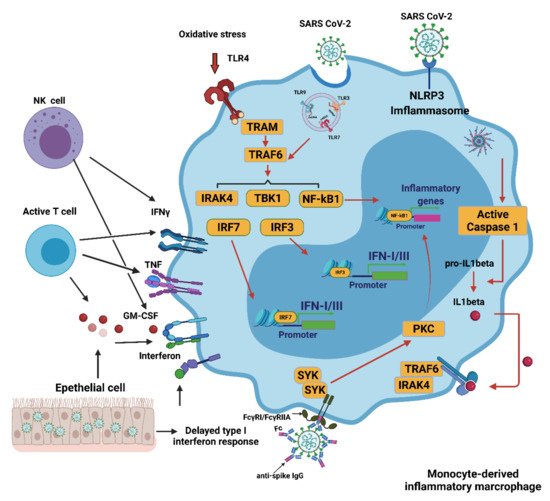
4.4. Contribution of Monocytes/Macrophages to Oxidative Stress
5. Adaptive Immunity in COVID-19
6. Aging in COVID-19
|
Cell Type/Signaling |
Aging |
Absence/Reduction in Type I IFN |
|---|---|---|
|
PRR activation & signaling |
↑ High age-related basal PRR (TLR) activation leads to excessive pro-inflammatory cytokine production ↓ Post-PRR downstream signaling activation |
↓ Recognition of intracellular pathogens ↑ Activity of Nlrp3 inflammasome ↓ ISGs expression signaling ↓Inducible nitric oxide synthase (iNOS) |
|
Neutrophils |
↑ Neutrophil influx through IL-17, CXCL1, CXCL2 ↓ Phagocytic function ↓ Signaling pathway |
↑ Neutrophil recruitment by CXCL1 and CXCL2 production by monocytes |
|
Monocyte/Macrophages |
↑ IL-6 and TNF production ↓ Macrophage phagocytosis of apoptotic neutrophils ↓ Alveolar macrophage affects repair of lung damage |
↓ Inflammatory monocyte-derived macrophages (IM) recruitment ↑ Resident IM proliferation ↓ IM iNOS, ↑ Ly6Clo monocyte iNOS production ↓ IM TRAIL expression ↓ Macrophage phagocytosis and efferocytosis of apoptotic neutrophils |
|
NK cells, Type I IFN |
↓ Delayed type I IFN activation and production ↓ Cytotoxic early viral clearance ↑ Cytokine production and consequent lung damage ↑ NK cell apoptosis |
↓ NK cell activation and IFN-production ↓ NK cell survival |
|
Dendritic cells |
↓ Functional capability ↓ DC maturation and migration to lymphoid organs affect T cell activation |
↑ cDC2 subtype development ↓ Antiviral responses by lowering ISGs ↓ Migration function |
↓: decrease; ↑: increase.
7. Resveratrol and COVID-19
7.1. Anti-Aging Interventions
7.2. Resveratrol as a Modulator of Antiviral Agent
References
- Gallo Marin, B.; Aghagoli, G.; Lavine, K.; Yang, L.; Siff, E.J.; Chiang, S.S.; Salazar-Mather, T.P.; Dumenco, L.; Savaria, M.C.; Aung, S.N.; et al. Predictors of COVID-19 severity: A literature review. Rev. Med. Virol. 2021, 31, 1–10.
- Feng, E.; Balint, E.; Poznanski, S.M.; Ashkar, A.A.; Loeb, M. Aging and Interferons: Impacts on Inflammation and Viral Disease Outcomes. Cells 2021, 10, 708.
- Marinella, M.A. Indomethacin and resveratrol as potential treatment adjuncts for SARS-CoV-2/COVID-19. Int. J. Clin. Pract. 2020, 74, e13535.
- Zhao, X.; Tong, W.; Song, X.; Jia, R.; Li, L.; Zou, Y.; He, C.; Liang, X.; Lv, C.; Jing, B.; et al. Antiviral Effect of Resveratrol in Piglets Infected with Virulent Pseudorabies Virus. Viruses 2018, 10, 457.
- Campagna, M.; Rivas, C. Antiviral activity of resveratrol. Biochem. Soc. Trans. 2010, 38, 50–53.
- Abba, Y.; Hassim, H.; Hamzah, H.; Noordin, M.M. Antiviral Activity of Resveratrol against Human and Animal Viruses. Adv. Virol. 2015, 2015, 184241.
- Acharya, D.; Liu, G.; Gack, M.U. Dysregulation of type I interferon responses in COVID-19. Nat. Rev. Immunol. 2020, 20, 397–398.
- Satarker, S.; Tom, A.A.; Shaji, R.A.; Alosious, A.; Luvis, M.; Nampoothiri, M. JAK-STAT Pathway Inhibition and their Implications in COVID-19 Therapy. Postgrad. Med. 2021, 133, 489–507.
- Gustine, J.N.; Jones, D. Immunopathology of Hyperinflammation in COVID-19. Am. J. Pathol. 2021, 191, 4–17.
- Matsuyama, T.; Kubli, S.P.; Yoshinaga, S.K.; Pfeffer, K.; Mak, T.W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020, 27, 3209–3225.
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetite, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129.
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518.
- Bournazos, S.; Gupta, A.; Ravetch, J.V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 2020, 20, 633–643.
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362.
- Delgado-Roche, L.; Mesta, F. Oxidative Stress as Key Player in Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) Infection. Arch. Med. Res. 2020, 51, 384–387.
- Cecchini, R.; Cecchini, A.L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses 2020, 143, 110102.
- Gan, R.; Rosoman, N.P.; Henshaw, D.J.E.; Noble, E.P.; Georgius, P.; Sommerfeld, N. COVID-19 as a viral functional ACE2 deficiency disorder with ACE2 related multi-organ disease. Med. Hypotheses 2020, 144, 110024.
- Fu, J.; Kong, J.; Wang, W.; Wu, M.; Yao, L.; Wang, Z.; Jin, J.; Wu, D.; Yu, X. The clinical implication of dynamic neutrophil to lymphocyte ratio and D-dimer in COVID-19: A retrospective study in Suzhou China. Thromb. Res. 2020, 192, 3–8.
- Rabelo, L.A.; Alenina, N.; Bader, M. ACE2-angiotensin-(1-7)-Mas axis and oxidative stress in cardiovascular disease. Hypertens. Res. 2011, 34, 154–160.
- Zablocki, D.; Sadoshima, J. Angiotensin II and oxidative stress in the failing heart. Antioxid. Redox Signal. 2013, 19, 1095–1109.
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-induced production of mitochondrial reactive oxygen species: Potential mechanisms and relevance for cardiovascular disease. Antioxid. Redox Signal. 2013, 19, 1085–1094.
- Rincon, J.; Correia, D.; Arcaya, J.L.; Finol, E.; Fernandez, A.; Perez, M.; Yaguas, K.; Talavera, E.; Chavez, M.; Summer, R.; et al. Role of Angiotensin II type 1 receptor on renal NAD(P)H oxidase, oxidative stress and inflammation in nitric oxide inhibition induced-hypertension. Life Sci. 2015, 124, 81–90.
- Valente, A.J.; Yoshida, T.; Murthy, S.N.; Sakamuri, S.S.; Katsuyama, M.; Clark, R.A.; Delafontaine, P.; Chandrasekar, B. Angiotensin II enhances AT1-Nox1 binding and stimulates arterial smooth muscle cell migration and proliferation through AT1, Nox1, and interleukin-18. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H282–H296.
- Sawalha, A.H.; Zhao, M.; Coit, P.; Lu, Q. Epigenetic dysregulation of ACE2 and interferon-regulated genes might suggest increased COVID-19 susceptibility and severity in lupus patients. Clin. Immunol. 2020, 215, 108410.
- Akbar, A.N.; Gilroy, D.W. Aging immunity may exacerbate COVID-19. Science 2020, 369, 256–257.
- Mueller, A.L.; McNamara, M.S.; Sinclair, D.A. Why does COVID-19 disproportionately affect older people? Aging (Albany NY) 2020, 12, 9959–9981.
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50.
- Schonrich, G.; Raftery, M.J.; Samstag, Y. Devilishly radical NETwork in COVID-19: Oxidative stress, neutrophil extracellular traps (NETs), and T cell suppression. Adv. Biol. Regul. 2020, 77, 100741.
- Violi, F.; Oliva, A.; Cangemi, R.; Ceccarelli, G.; Pignatelli, P.; Carnevale, R.; Cammisotto, V.; Lichtner, M.; Alessandri, F.; De Angelis, M.; et al. Nox2 activation in COVID-19. Redox Biol. 2020, 36, 101655.
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513.
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062.
- Cavezzi, A.; Troiani, E.; Corrao, S. COVID-19: Hemoglobin, iron, and hypoxia beyond inflammation. A narrative review. Clin. Pract. 2020, 10, 1271.
- Iwasaki, A.; Foxman, E.F.; Molony, R.D. Early local immune defences in the respiratory tract. Nat. Rev. Immunol. 2017, 17, 7–20.
- Guo, H.Y.; Zhang, X.C.; Jia, R.Y. Toll-Like Receptors and RIG-I-Like Receptors Play Important Roles in Resisting Flavivirus. J. Immunol. Res. 2018, 2018, 6106582.
- Chow, K.T.; Gale, M., Jr.; Loo, Y.M. RIG-I and Other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol. 2018, 36, 667–694.
- Barrat, F.J.; Elkon, K.B.; Fitzgerald, K.A. Importance of Nucleic Acid Recognition in Inflammation and Autoimmunity. Annu. Rev. Med. 2016, 67, 323–336.
- Kindler, E.; Thiel, V.; Weber, F. Interaction of SARS and MERS Coronaviruses with the Antiviral Interferon Response. Adv. Virus Res. 2016, 96, 219–243.
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245.
- Tay, M.Z.; Poh, C.M.; Renia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374.
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923.
- Supramaniam, A.; Lui, H.; Bellette, B.M.; Rudd, P.A.; Herrero, L.J. How myeloid cells contribute to the pathogenesis of prominent emerging zoonotic diseases. J. Gen. Virol. 2018, 99, 953–969.
- Wack, A.; Terczynska-Dyla, E.; Hartmann, R. Guarding the frontiers: The biology of type III interferons. Nat. Immunol. 2015, 16, 802–809.
- Broggi, A.; Ghosh, S.; Sposito, B.; Spreafico, R.; Balzarini, F.; Lo Cascio, A.; Clementi, N.; De Santis, M.; Mancini, N.; Granucci, F.; et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science 2020, 369, 706–712.
- Zhou, Z.; Ren, L.; Zhang, L.; Zhong, J.; Xiao, Y.; Jia, Z.; Guo, L.; Yang, J.; Wang, C.; Jiang, S.; et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe 2020, 27, 883–890e2.
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045e9.
- Chu, H.; Chan, J.F.; Wang, Y.; Yuen, T.T.; Chai, Y.; Hou, Y.; Shuai, H.; Yang, D.; Hu, B.; Huang, X.; et al. Comparative Replication and Immune Activation Profiles of SARS-CoV-2 and SARS-CoV in Human Lungs: An Ex Vivo Study with Implications for the Pathogenesis of COVID-19. Clin. Infect. Dis. 2020, 71, 1400–1409.
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Pere, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724.
- Garcia-Del-Barco, D.; Risco-Acevedo, D.; Berlanga-Acosta, J.; Martos-Benitez, F.D.; Guillen-Nieto, G. Revisiting Pleiotropic Effects of Type I Interferons: Rationale for Its Prophylactic and Therapeutic Use against SARS-CoV-2. Front. Immunol. 2021, 12, 655528.
- Hackbart, M.; Deng, X.; Baker, S.C. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc. Natl. Acad. Sci. USA 2020, 117, 8094–8103.
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468.
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 2020, 369, 1249–1255.
- Freitas, B.T.; Durie, I.A.; Murray, J.; Longo, J.E.; Miller, H.C.; Crich, D.; Hogan, R.J.; Tripp, R.A.; Pegan, S.D. Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect. Dis. 2020, 6, 2099–2109.
- Frieman, M.; Baric, R. Mechanisms of severe acute respiratory syndrome pathogenesis and innate immunomodulation. Microbiol. Mol. Biol. Rev. 2008, 72, 672–685, Table of Contents.
- Pauli, E.K.; Schmolke, M.; Wolff, T.; Viemann, D.; Roth, J.; Bode, J.G.; Ludwig, S. Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog. 2008, 4, e1000196.
- Smits, S.L.; de Lang, A.; van den Brand, J.M.; Leijten, L.M.; van, I.W.F.; Eijkemans, M.J.; van Amerongen, G.; Kuiken, T.; Andeweg, A.C.; Osterhaus, A.D.; et al. Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathog. 2010, 6, e1000756.
- Lee, Y.; Shin, H.; Kim, J. In vivo Anti-Cancer Effects of Resveratrol Mediated by NK Cell Activation. J. Innate Immun. 2021, 13, 94–106.
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Dassler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652.
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147.
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691.
- Raup-Konsavage, W.M.; Wang, Y.; Wang, W.W.; Feliers, D.; Ruan, H.; Reeves, W.B. Neutrophil peptidyl arginine deiminase-4 has a pivotal role in ischemia/reperfusion-induced acute kidney injury. Kidney Int. 2018, 93, 365–374.
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6676.
- Schonrich, G.; Raftery, M.J. Neutrophil Extracellular Traps Go Viral. Front. Immunol. 2016, 7, 366.
- Sil, P.; Wicklum, H.; Surell, C.; Rada, B. Macrophage-derived IL-1beta enhances monosodium urate crystal-triggered NET formation. Inflamm. Res. 2017, 66, 227–237.
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320.
- Papayannopoulos, V.; Staab, D.; Zychlinsky, A. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PLoS ONE 2011, 6, e28526.
- Kim, O.Y.; Monsel, A.; Bertrand, M.; Coriat, P.; Cavaillon, J.M.; Adib-Conquy, M. Differential down-regulation of HLA-DR on monocyte subpopulations during systemic inflammation. Crit. Care 2010, 14, R61.
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501e15.
- Nikolich-Zugich, J. The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 2018, 19, 10–19.
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients with Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827.
- Goronzy, J.J.; Weyand, C.M. Successful and Maladaptive T Cell Aging. Immunity 2017, 46, 364–378.
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020, 46, 846–848.
- Nikolich-Zugich, J.; Knox, K.S.; Rios, C.T.; Natt, B.; Bhattacharya, D.; Fain, M.J. SARS-CoV-2 and COVID-19 in older adults: What we may expect regarding pathogenesis, immune responses, and outcomes. Geroscience 2020, 42, 505–514.
- Koelman, L.; Pivovarova-Ramich, O.; Pfeiffer, A.F.H.; Grune, T.; Aleksandrova, K. Cytokines for evaluation of chronic inflammatory status in ageing research: Reliability and phenotypic characterisation. Immun. Ageing 2019, 16, 11.
- Xia, S.; Zhang, X.; Zheng, S.; Khanabdali, R.; Kalionis, B.; Wu, J.; Wan, W.; Tai, X. An Update on Inflamm-Aging: Mechanisms, Prevention, and Treatment. J. Immunol. Res. 2016, 2016, 8426874.
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217.
- Chung, H.Y.; Sung, B.; Jung, K.J.; Zou, Y.; Yu, B.P. The molecular inflammatory process in aging. Antioxid. Redox Signal. 2006, 8, 572–581.
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol. Immunol. 2020, 17, 533–535.
- Park, M.D. Macrophages: A Trojan horse in COVID-19? Nat. Rev. Immunol. 2020, 20, 351.
- Wang, W.; Xu, Y.; Gao, R.; Lu, R.; Han, K.; Wu, G.; Tan, W. Detection of SARS-CoV-2 in Different Types of Clinical Specimens. JAMA 2020, 323, 1843–1844.
- Bajaj, V.; Gadi, N.; Spihlman, A.P.; Wu, S.C.; Choi, C.H.; Moulton, V.R. Aging, Immunity, and COVID-19: How Age Influences the Host Immune Response to Coronavirus Infections? Front. Physiol. 2020, 11, 571416.
- Blagosklonny, M.V. from causes of aging to death from COVID-19. Aging (Albany NY) 2020, 12, 10004–10021.
- Li, Y.R.; Li, S.; Lin, C.C. Effect of resveratrol and pterostilbene on aging and longevity. BioFactors 2018, 44, 69–82.
- Sieck, G.C. Physiology in Perspective: Understanding the Aging Process. Physiology 2018, 33, 372–373.
- Meftahi, G.H.; Jangravi, Z.; Sahraei, H.; Bahari, Z. The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID-19 infection: The contribution of "inflame-aging". Inflamm. Res. 2020, 69, 825–839.
- Colantuoni, A.; Martini, R.; Caprari, P.; Ballestri, M.; Capecchi, P.L.; Gnasso, A.; Lo Presti, R.; Marcoccia, A.; Rossi, M.; Caimi, G. COVID-19 Sepsis and Microcirculation Dysfunction. Front. Physiol. 2020, 11, 747.
- Zhang, B.; Zhou, X.; Qiu, Y.; Song, Y.; Feng, F.; Feng, J.; Song, Q.; Jia, Q.; Wang, J. Clinical characteristics of 82 cases of death from COVID-19. PLoS ONE 2020, 15, e0235458.
- Kaleta, E.F. Herpesviruses of birds—A review. Avian Pathol. 1990, 19, 193–211.
- Zhao, X.; Xu, J.; Song, X.; Jia, R.; Yin, Z.; Cheng, A.; Jia, R.; Zou, Y.; Li, L.; Yin, L.; et al. Antiviral effect of resveratrol in ducklings infected with virulent duck enteritis virus. Antivir. Res. 2016, 130, 93–100.
- Parashar, U.D.; Hummelman, E.G.; Bresee, J.S.; Miller, M.A.; Glass, R.I. Global illness and deaths caused by rotavirus disease in children. Emerg. Infect. Dis. 2003, 9, 565–572.
- Cui, Q.; Fu, Q.; Zhao, X.; Song, X.; Yu, J.; Yang, Y.; Sun, K.; Bai, L.; Tian, Y.; Chen, S.; et al. Protective effects and immunomodulation on piglets infected with rotavirus following resveratrol supplementation. PLoS ONE 2018, 13, e0192692.
- Baldassarre, M.E.; Di Mauro, A.; Labellarte, G.; Pignatelli, M.; Fanelli, M.; Schiavi, E.; Mastromarino, P.; Capozza, M.; Panza, R.; Laforgia, N. Resveratrol plus carboxymethyl-β-glucan in infants with common cold: A randomized double-blind trial. Heliyon 2020, 6, e03814.
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820.
- Lin, S.C.; Ho, C.T.; Chuo, W.H.; Li, S.; Wang, T.T.; Lin, C.C. Effective inhibition of MERS-CoV infection by resveratrol. BMC Infect. Dis. 2017, 17, 144.
- Jiang, Z.; Zhang, H.; Gao, J.; Yu, H.; Han, R.; Zhu, L.; Chen, X.; Fan, Q.; Hao, P.; Wang, L.; et al. ACE2 Expression Is Upregulated in Inflammatory Corneal Epithelial Cells and Attenuated by Resveratrol. Investig. Ophthalmol. Vis. Sci. 2021, 62, 25.
- Perrella, F.; Coppola, F.; Petrone, A.; Platella, C.; Montesarchio, D.; Stringaro, A.; Ravagnan, G.; Fuggetta, M.P.; Rega, N.; Musumeci, D. Interference of Polydatin/Resveratrol in the ACE2:Spike Recognition during COVID-19 Infection. A Focus on Their Potential Mechanism of Action through Computational and Biochemical Assays. Biomolecules 2021, 11, 1048.
- Ramdani, L.H.; Bachari, K. Potential therapeutic effects of Resveratrol against SARS-CoV-2. Acta Virol. 2020, 64, 276–280.
- Yang, L.; Wang, Z. Natural Products, Alone or in Combination with FDA-Approved Drugs, to Treat COVID-19 and Lung Cancer. Biomedicines 2021, 9, 689.
- Rossi, G.A.; Sacco, O.; Capizzi, A.; Mastromarino, P. Can Resveratrol-Inhaled Formulations Be Considered Potential Adjunct Treatments for COVID-19? Front. Immunol. 2021, 12, 670955.
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687.

