 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Melkam Alamerew Kebede | + 4878 word(s) | 4878 | 2021-08-13 11:48:38 | | | |
| 2 | Conner Chen | -302 word(s) | 4576 | 2021-09-13 06:21:31 | | |
Video Upload Options
The pancreatic β-cell is purpose-built for the production and secretion of insulin, the only hormone that can remove glucose from the bloodstream. Insulin is kept inside miniature membrane-bound storage compartments known as secretory granules (SGs), and these specialized organelles can readily fuse with the plasma membrane upon cellular stimulation to release insulin. The luminal components of the insulin SG can be functionally segregated into four groups. These are cargo molecules, luminal enzymes and chaperones, ions (and their transporters and channels), and sorting receptors.
1. Introduction
The insulin secretory granule (SG) in the pancreatic β-cell is essential for glucose homeostasis in the body. It is both the site of proinsulin conversion into insulin and C-peptide [1], as well as the storage compartment for mature insulin to be readily available for secretion upon nutrient stimuli. Insulin is first synthesized as pre-proinsulin at the endoplasmic reticulum (ER), immediately converted to proinsulin, and transported through the Golgi to the trans-Golgi network (TGN). Here, proinsulin, along with other cargo proteins, is partitioned and sorted into its destination compartment, the immature SG (ISG) [1]. In the ISG, at least 99% of proinsulin is ultimately converted to insulin and C-peptide in a 1:1 molar ratio via proteolytic cleavages by the proprotein convertases PC1/3 and PC2 [2][3][4][5]. This coincides with several processes that facilitate SG maturation, including luminal acidification [6], selective removal of certain soluble components [7], and Zn2+-mediated insulin crystallization [8]. Finally, in response to nutrient stimuli, these mature SGs (MSGs) are mobilized to fuse with the plasma membrane and deliver insulin to the bloodstream.
Importantly, ISGs can also undergo regulated secretion [9], which can be heightened in situations of increased β-cell demand [10][11][12] and may explain the higher circulating proinsulin to insulin ratio observed in both pre-diabetic and diabetic patients [13][14][15][16][17][18][19]. The mechanism behind increased proinsulin secretion is unknown; although it has been suggested to result from defective proinsulin trafficking or processing, and/or the premature release of ISGs [20][21]. Interestingly, β-cells from animal models of type 2 diabetes (T2D) display a compensatory expansion of the secretory pathway, characterized by increased proinsulin biogenesis but exhibit a thorough depletion of MSGs, pointing to the existence of a bottleneck in the secretory pathway resulting in an MSG replenishment defect during β-cell failure [12]. Therefore, there is a diversion away from SG maturation in favor of ISG secretion, limiting the compensatory capacity of the β-cell during metabolic stress.
Alongside insulin, the β-cell SG contains a cocktail of cargo proteins. These proteins drive trafficking through the regulated secretory pathway and are also released to affect systemic function [22][23][24][25]. Luminal enzymes accompany the cargo from synthesis in the ER through to storage in the MSG but are under tight regulation to restrict their activity to the correct site [4]. The ionic composition of the lumen controls protein behavior and is generated by a range of transmembrane channels and transporters that are stationed throughout the secretory pathway [26][27][28][29][30][31]. Finally, sorting receptors can escort unwanted components away from the maturing SG to refine its contents after formation [7][32].
2. Cargo Molecules
The primary cargoes of SGs in the pancreatic β-cell are insulin, islet amyloid polypeptide (IAPP), the granins [chromogranin A (CgA), chromogranin B (CgB), secretogranin II (SgII), secretogranin III (SgIII), and VGF (non-acronymic)], and each of their precursors and derivatives. In addition to those covered in this review, the insulin SG also contains amines such as dopamine and serotonin [33][34][35], as well as nucleotides like ATP [36], which can be taken up by SG-localized pumps but as of yet, have ill-defined intragranular and post-exocytotic roles [37].
Insulin. Insulin is synthesized as pre-proinsulin on the rough ER, and upon translocation has its N-terminal 24-residue signal sequence cleaved to form proinsulin [38]. Proinsulin undergoes folding in the ER where it acquires three disulfide bonds and dimerizes prior to ER exit [39][40]. En route to the TGN, proinsulin forms hexamers in the presence of Zn2+ [39][41], and importantly, proinsulin hexamers remain soluble [42]. Zn2+ binds to a histidine corresponding to residue 10 on the B chain of mature insulin (His-B10), and while the precise cisternal location of this event is undetermined, there is evidence of a Zn2+-dependent rate limiting step for proinsulin trafficking around the TGN/ISG compartment [40].
After entry into the ISG, proinsulin is converted to insulin and C-peptide via ordered cleavage at two sites of dibasic amino acid residues by the subtilisin-related proprotein convertases, first by PC1/3 and then by PC2 (Figure 1A). The 31–32 Arg-Arg site is located at the C-peptide/B-chain junction and the 64–65 Lys-Arg site is located between the C-peptide/A-chain junction. Molecular modelling suggests that the co-ordination of Zn2+ by His-B10 works to position these sites along the exposed radial surface of the proinsulin hexamer [43], enabling accessibility for the two processing enzymes. PC1/3 preferentially cleaves the B-chain junction on the carboxyl side of Arg32, generating a proinsulin intermediate split between residues 32 and 33 (split 32,33 proinsulin) [4][5][44]. PC2 preferentially cleaves the A-chain junction on the carboxyl side of Arg65 to generate the split 65,66 proinsulin intermediate [4][5][44]. Following conversion by each of the subtilisin-related prohormone convertases, the exoprotease carboxypeptidase H/E (CPE) acts to trim the revealed dibasic residues to create the ‘des’ intermediates, des 31, 32, or des 64,65 proinsulin, with numbers denoting the excised residues [45]. A second round of endoprotease and CPE activity will generate insulin and C-peptide in a 1:1 molar ratio [3][4]. The insulin molecule consists of an A-chain and a B-chain, linked together by two disulfide bridges and maintained in hexameric oligomers through the co-ordination of two-Zn2+ by three of the six His-B10s [8][46]. Continual uptake of H+ and Zn2+ into the developing SG affects the charge state of hexameric insulin and facilitates its packing into extremely insoluble crystals [47]. The low percentage of unprocessed/incompletely processed proinsulin can pack with crystalline insulin to some extent [48] and C-peptide can co-precipitate with insulin in pH conditions mimicking the MSG [49]. C-peptide can also undergo further exoproteolytic cleavage to generate des 27–31 C-peptide, accounting for roughly 10% of the total C-peptide content [50]. Upon exocytosis, exposure to the neutral extracellular pH is likely to dissipate the insulin crystal rapidly [51], allowing monomeric insulin to circulate and signal via the insulin receptor expressed on target tissues.
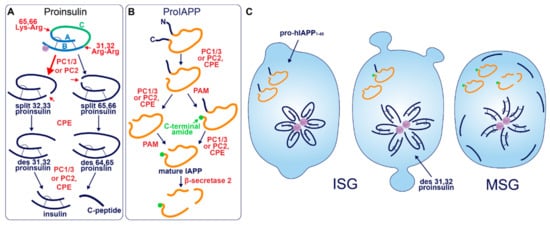
Figure 1. Prohormone Processing in the β-cell. (A) Sequence of proinsulin processing. After entry into the ISG, proinsulin is converted to insulin and C-peptide via cleavage at two sites of dibasic amino acid residues. The 31–32 Arg-Arg site is located at the C-peptide/B-chain junction and the 64–65 Lys-Arg site is located between the C-peptide/A-chain junction. Cleavage at one dibasic site by endoprotease PC1/3 or PC2 produces the split proinsulin molecules, which precedes C-terminal trimming of exposed residues by exoprotease CPE to produce the des proinsulin molecules. One round of endo/exoprotease activity is followed by the same action at the other dibasic site. (B) Sequence of proIAPP processing. The C-terminal proregion of proIAPP is cleaved in the TGN prior to ISG entry. Next, in no particular order, within the maturing ISG the N-terminal proregion of proIAPP is removed and the exposed C-terminal glycine residue is amidated to produce IAPP. IAPP may then be further processed into smaller fragments by β-secretase 2. (C) Processing events and products during secretory granule maturation in the human β-cell SG. des 31,32 proinsulin is the major proinsulin intermediate in human β-cells and is elevated in the circulation of those with T2D along with proIAPP1–48.
A human mutation of His-B10 to aspartate (mAsp-B10) underlies familial hyperproinsulinemia [52] and represents a condition where mutant proinsulin is presumed to be excluded from wild-type proinsulin hexamers. While expression of this mutant in mice does not affect its intracellular conversion to insulin, there is an enrichment of non-crystallized SGs, and the constitutive release of proinsulin is increased by ~15% [53]. These phenotypes could indicate that mAsp-B10 proinsulin is correctly targeted into the ISG, but there is an increased constitutive-like release in the absence of Zn2+-facilitated hexamarization prior to its conversion into insulin. Indeed, while contributing to the maturation of the SG, constitutive-like secretion is estimated to account for only 0.6% of the release of non-converted proinsulin [54]. This situation could represent an extreme example of protein exit out of the ISG, displaying the secretory capacity of the constitutive-like pathway. An alternative (and not mutually exclusive) explanation is that mAsp-B10 proinsulin leaks directly into the constitutive pathway from the TGN, however mAsp-B10 proinsulin degradation is also enhanced [53] suggesting that its transit to the PM occurs through the constitutive-like pathway (a route that travels via the endo-lysosomal system [42]). Nonetheless, these studies have highlighted that Zn2+-facilitated hexamarization is a primary mechanism of proinsulin sorting and consequent SG maturation.
Early studies investigating human proinsulin and rat proinsulin isomers I and II in primary islets revealed that they are differentially processed. Human proinsulin tends to be cleaved first at the B-chain junction to produce des 31,32 proinsulin [55] (Figure 1C), whereas the rat isomers tend to be cleaved first at the A-chain junction to produce des 64,65 proinsulin [56]. This is thought to be due to the amino acid located four residues prior to the cleavage site (P4 position [57]), where the presence of a basic lysine or arginine residue enhances substrate recognition and/or enzymatic activity [58]. Both rat isomers contain a basic arginine at P4 in the A-chain site, and both rat proinsulin I and human proinsulin contain a basic lysine at P4 in the B-chain site [59]. As a result, rat proinsulin I is more rapidly converted into insulin, and the accumulation of processing intermediates from this isomer is reduced due to the existence of basic residues at P4 in both cleavage sites [56].
Processing of human proinsulin follows a sequence favoring the prior activity of PC1/3 on the B-chain junction, followed by PC2, which has a far better affinity for des 31,32 proinsulin than intact proinsulin [60]. Although this points to the existence of sequential cleavage through the action of both endoproteases, multiple lines of evidence indicate that PC1/3 works alone to produce mature insulin from both rat and human proinsulin isomers. While each enzyme possesses the catalytic ability to cleave at both dibasic sites [61], PC1/3 achieves this far more efficiently than PC2 [62][63][64] and processing intermediates of proinsulin accumulate when PC1/3 expression is low [63]. The situation is different in mice, seemingly requiring the activity of both endoproteases; while the deletion of PC1/3 from mice results in an extremely pronounced block in proinsulin conversion [65], knockout of PC2 also significantly hampers insulin maturation despite the presence of PC1/3 [66]. Finally, a recent study that re-characterized the expression of PC1/3 and PC2 in human islet β-cells found an abundance of PC1/3 and an absence of PC2, suggesting that PC1/3 is sufficient for humans to produce insulin [67]. Interestingly, humans with T2D had upregulated PC1/3 and an induction of PC2 expression. The authors of this study speculated that aberrant PC2 expression could cause a processing defect that underlies the pathological state, although, it may be the case that PC2 expression is invoked by metabolic stress as a compensatory response to assist PC1/3 in proteolytic activities. Indeed, the catalytic rate of PC2 on des 31,32 proinsulin exceeds that of PC1/3 on intact proinsulin [60]. Simple overexpression of either PC1/3 or PC2 has been shown to enhance proinsulin conversion in rat insulinoma INS1 cells [68], hence, induction of PC2 activity could support proinsulin conversion when PC1/3 is overwhelmed especially considering that the A-chain junction is not preferred by PC1/3 [4][44].
Both PC1/3 and PC2 endoprotease activities are sensitive to pH and Ca2+. In vitro assays using enzymes isolated from rat islets have shown that PC1/3 requires millimolar levels of Ca2+ and a pH close to 5.5 for activity whereas PC2 can exert activity at a micromolar levels of Ca2+ and over a broader pH range, although its pH optimum is also 5.5 [4]. In cells however, PC1/3 undergoes fast maturation into an active enzyme upon entry into the SG [69]. Due to the stringent regulation of PC2 by the molecular chaperone 7B2 [70][71][72], the low pH requirement for its autocatalytic activation [69][73], as well as its substrate-specificity to des 31,32 proinsulin [60], its activity is likely to be restricted to later stages of SG maturation. Therefore, it appears that early PC1/3 activity at both sites could render PC2 redundant, as has been demonstrated in animal models [62][63][64] but not quite yet in humans. Crucially, compensatory upregulation of the endoproteases may be futile, considering the premature ISG release that occurs in β-cell failure. It has been known for some time that des 31,32 intermediates are the predominant species of circulating proinsulin that is elevated in human T2D [74][75], therefore fast endoprotease activity is critical for systemic metabolic homeostasis. Therapeutic compounds that alter the ionic composition of the SG to bolster endoprotease activation and activity could be effective in treating T2D.
Despite a common outcome, nuances in the generation of insulin are clear between species. Their awareness may be important for translating data from model organisms to the context of human β-cell function.
Islet Amyloid Polypeptide. IAPP is a 37 amino-acid peptide stored in the MSG that is co-secreted with insulin in a 1:100 molar ratio [76][77][78], and can function to suppress insulin secretion and control various aspects of energy homeostasis [22][79]. Additionally, known as amylin, IAPP and its precursors and derivatives are notorious for forming fibrils that distribute extracellularly throughout islets as amyloid deposits, a pathological feature of human T2D [80]. Early observations report the occurrence of islet amyloid deposits in >90% of diabetic patients [81][82] but later studies have shown a variable prevalence depending on duration of disease and ethnicity, especially when sample size is increased [83]. The question of how IAPP remains non-pathogenic in healthy conditions and how it transitions to a pathogenic molecule has kept researchers occupied for some time. Appropriately, most work on IAPP has been focused on its secretory dynamics, processing, and amyloidogenic properties [84][85][86] rather than its luminal sorting. To this end, not much is known about its behavior in the early secretory pathway or the determinants of its trafficking fate.
Akin to proinsulin, human pro-IAPP is a 67 amino-acid (aa) peptide derived from pre-proIAPP that forms an intramolecular disulfide bridge in the ER and is subject to endoproteolytic processing [87][88][89][90] (Figure 1B). It is thought that PC1/3 acts first on a C-terminal proregion in the TGN [89] which is followed by CPE action to generate a 48-residue processing intermediate. A subsequent round of PC1/3 or PC2 and CPE action on the N-terminal proregion generates a 38 aa peptide with a C-terminal Gly termed amylin free acid [91]. A fourth enzyme, peptidyl-glycine alpha-amidating monooxygenase (PAM), is probably responsible for amidation at the C-terminal 38 glycine residue which may or may not occur prior to cleavage of the N-terminal pro-region, to generate the mature C-terminally amidated form of IAPP [91]. Finally, a fifth membrane-bound enzyme that localizes to the β-cell SG, β-secretase 2, can process IAPP further into smaller fragments [92].
Early reports demonstrated that PC2 can cleave at both the N- and C-terminal proregions of pro-mouse IAPP (mIAPP) in addition to PC1/3 cleavage occurring only at the C-terminal site [87][88][89][90]. However, both rat and human islets appear not to express PC2 at detectable levels normally [67]. Indeed, pro-human IAPP (hIAPP) can be fully processed by PC1/3 in PC2 null mice [93]; however, pro-rat IAPP (rIAPP) can be processed at both sites by PC2 but only at one site by PC1/3 [90]. These differences are likely due to the modified sequence at the C-terminal site (Figure 2), where the position of Ala and Val residues may determine whether PC1/3 can cleave: i.e., KR↓VA (Val at P1′ and Ala at P2′) in rIAPP compared to KR↓AV (Ala at P1′ and Val at P2′) in hIAPP and KR↓AA (Ala at P1′ and P2′) in mIAPP. Likewise, it has also been suggested that a Val common to both hIAPP and rIAPP at the N-terminal site allows cleavage by PC1/3 [67], which is not experienced by mIAPP [87] that contains Met at this residue. Thus, PC1/3 may be sufficient for full pro-IAPP processing in humans due to the sequence variations that lie between species.

Figure 2. IAPP precursor/product amino acid sequence in human (H), mouse (M), and rat (R). The green glycine residue is amidated after the C-terminal cleavage site is processed. Red residues denote dibasic sites of endoproteolytic processing. Blue residues indicate a modified sequence between species at cleavage sites that could account for their differential specificity to PC enzymes.
IAPP resides in the soluble fraction of the MSG, and hIAPP is extremely fibrillogenic whereas rIAPP and mIAPP are not fibrillogenic at all [94]. It has been shown in vitro that pro-hIAPP products become increasingly more amyloidogenic with further cleavages [95], however, it is thought that the presence of ionic and molecular factors in the MSG periphery inhibits hIAPP oligomer formation in healthy conditions [96][97][98][99][100][101]. An extensive body of literature has covered the molecular mechanisms of hIAPP pathogenicity in T2D [84][85][86]. Considering that hIAPP is likely not a driver of SG biogenesis or function but instead plays the role of chaotic passenger, we will focus here on how secretory pathway dysfunction could precede hIAPP-mediated β-cell damage.
A current working hypothesis to explain the initiation of islet amyloid formation is that hIAPP-related peptides undergo dysregulated fibrillogenesis at some point inside the β-cell secretory pathway, potentially due to overproduction during compensation/failure [102], but this only occurs within a small subset of islet β-cells [84][85][103][104][105]. If pro-hIAPP overproduction is the fuel, then disruption of organellar membranes from within the cell is the spark that results in β-cell death and the deposition of extracellular amyloid. Through regular exocytosis from neighboring β-cells, released hIAPP can then add to the size of the initial deposit. An alternative (and not mutually exclusive) hypothesis suggests that release of the 48-residue pro-hIAPP (pro-hIAPP1–48) intermediate can initiate extracellular amyloid formation by a specific interaction with heparin sulfate proteoglycans in the extracellular matrix [106]. Interestingly, large ordered fibrils that make up the bulk of the visually identified IAPP deposition are thought to be relatively inert on a cytotoxic level [86], although interruption of islet cytoarchitecture could impair coordinated islet function. Rather, it is the presence of medium-sized disordered oligomers that are thought to exert most of the cellular damage [107]. Fitting with this, recent experimental focus has instead been placed on the mechanism of medium-sized oligomer formation and cytotoxicity [108][109].
Not surprisingly, the N-terminal prosequence of hIAPP is detectable in islet amyloid deposits [110][111]. This observation resembles what is observed with proinsulin in that incompletely processed hIAPP may be released from the β-cell during T2D, and indeed, elevated serum pro-hIAPP has been observed in glucose intolerant and T2D patients [112]. If one considers that ISGs released during β-cell failure are a source of hIAPP processing intermediates (Figure 1C), we could look to luminal factors that might explain the propensity of these molecules to become pathogenic. Indeed, insulin, Zn2+, H+, Ca2+, C-peptide, and proinsulin have been assessed individually or in combination, in vitro, along with hIAPP, in various studies to reason that a delicate balance of cofactors is required to inhibit hIAPP oligomerization [96][97][98][99][100][101][113]. In healthy cells, regulated exocytosis of MSGs could maintain this balance as components are released in an appropriate molar ratio. Conversely, during β-cell compensation and failure, release of the incompletely formed ISG might not replicate this outcome, and cytotoxic hIAPP oligomers could form in the extracellular microenvironment adjacent to the plasma membrane to induce membrane damage.
Dysregulated hIAPP oligomerization exacerbates the progression of T2D, so preventing β-cell death at the hands of hIAPP could limit T2D severity. Abnormal SG composition or the premature release of ISGs may be contributing factors, highlighting the importance of correctly forming the insulin SG.
Granins. The granin family of proteins (CgA, CgB, SgII, SgIII, and VGF) are ubiquitously expressed across neurons and endocrine cells and are considered major contributors to the biogenesis of regulated SGs from within the lumen (Figure 2). Their effectiveness has been displayed by several groups with findings that expression of just a single granin in cells that do not have a regulated secretory pathway is able to produce SGs that are capable of regulated release [24][114][115][116][117]. Granins are synthesized as soluble cargo precursor proteins, which are highly acidic and hydrophilic, but are prone to aggregation under mild acidity (pH < 6.4) and high Ca2+ (>1 mM) conditions [25][118]. It has been shown that both of these ionic requirements must be met for granin aggregation [118][119], which can be achieved at the initiating site of SG biogenesis in the TGN where ion pumps maintain a high luminal Ca2+ concentration and contribute to a substantial lowering of the pH through enhanced H+ uptake [27][120][121][122]. Moreover, several granins have been shown to interact with each other, and, lacking transmembrane domains themselves, some can also interact with lipid species on the luminal aspect of the secretory pathway membrane to provide a link between soluble and membrane fractions. In this way, their physical abundance, coordinated aggregation within the TGN, and binding to specific components on the membrane has been proposed to drive the segregation and sorting of peptide hormones and other proteins into the regulated secretory pathway [23], meeting the requirements of a ‘refined bulk-flow’ sorting by entry mechanism [123].
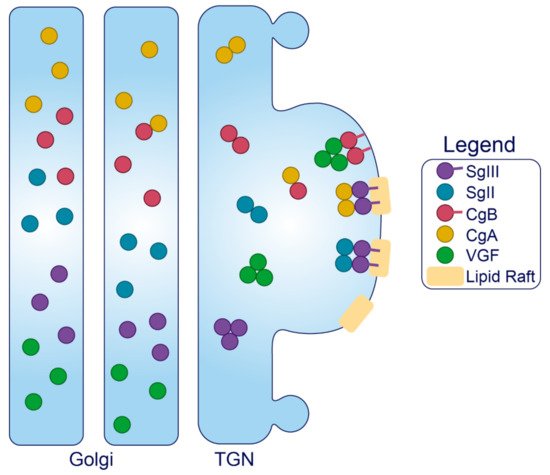
Figure 3. Trafficking of the granins. Upon exposure to mild acidity (pH < 6.4) within the Golgi apparatus, the granins will bind Ca2+, which triggers their aggregation and interaction with other granin members. In the TGN, several of these members will interact with target molecules in the membrane to drive the formation of SGs at distinct sites from within the lumen.
Sphingolipid–cholesterol lipid rafts accumulate in microdomains of the TGN [124][125], and these rafts can alter the distribution of transmembrane components to create sorting stations that are essential for granule biogenesis [126][127]. These rafts are also enriched in vesicles of the regulated secretory pathway [128][129][130][131], indicating that SG membranes originate from the sorting domains where granins and other SG constituents aggregate and bind. Importantly, saturated fatty acid and cholesterol intake can change the composition of lipid species distributed among cell membranes to influence trafficking and SG morphology [132][133]. Therefore, dietary status could affect interactions between the granins and secretory pathway membranes, but this requires investigation. Finally, since granins can bind Ca2+ at a high capacity with low affinity [119], they are also thought to equip the SG with the ability to store and release Ca2+ [134][135].
It should be noted that most of the literature on granins has reported on their role in neuronal/neuroendocrine cell lines, which although share features in common with the β-cell, have secretory pathways adapted to the specific needs of neural transmission. In these settings, we can draw insight from molecular interactions that govern trafficking and behavior of the granins themselves, but specific effects of granin depletion on SG biogenesis/secretion are often subject to cell-specific variation and thus will only be discussed with respect to the β-cell.
Granins can possess multiple sorting determinants and may be targeted to several SG sub-populations (Figure 3). SgIII is membrane-associated, and contains an N-terminal lipid-binding region that is required for its sorting into SGs of AtT-20 cells [136] and for its interaction with cholesterol in INS1 and AtT-20 SGs [137]. This suggests that SgIII is sorted into the regulated secretory pathway through an interaction with TGN cholesterol [136]. N-terminal residues (48–111) of CgA can bind to SgIII to follow SgIII sorting into the regulated secretory pathway of AtT-20 cells, where it also exists in association with SG membranes [137]. Importantly, CgA also associates with INS1 granule membranes but only in the presence of SgIII [136]. These results collectively indicate that SgIII is an adaptor for CgA in β-cells, with both granins associated at least to some degree with the SG membrane, and that correct trafficking relies on the presence of an N-terminal region on SgIII that binds cholesterol. This has been further demonstrated in PC12 cells, where SgIII was specifically shown to sort large aggregates of CgA into SGs [138]. Very little is known about the trafficking determinants of SgII aside from an understanding that both the N- and C-termini contain information for sorting [139], and that it may interact with SgIII on the SG membrane [140][141]. While SgII also regulates granule biogenesis in other secretory cells [142], interactions have not been published in β-cell models, and in general, insight into SgII function in the β-cell is lacking.
Like SgIII, CgB can interact with cellular membranes via a highly conserved N-terminal 22 residue disulfide-bonded loop [143][144]. This loop is essential for CgB sorting to SGs but is not required for its aggregation within the TGN, indicating that CgB aggregates are not routed by default to SGs but are sorted through mediation of exposed N-terminal loops with the TGN membrane [143]; although, the corresponding membrane component is yet to be found. Several observations suggest that CgB trafficking is not entirely synchronous with insulin. In addition to the insulin SG, CgB also occupies distinct granules that do not contain insulin and conversely, insulin can occupy SGs devoid of CgB [145]. Additionally, CgB is present with SgII in nucleoplasmic vesicles of bovine chromaffin cells where they may have a role in regulating nuclear Ca2+ homeostasis [146], although this has not been studied in the β-cell. In the β-cell, CgB co-localizes and co-immunoprecipitates with VGF [147], and it has been shown in vitro that CgA and CgB can form dimers at pH 7.5 and heterotetramers at pH 5.5 [148], suggesting that CgB could traffic with either VGF or CgA. However, VGF does not immunoprecipitate with CgA [147]. Little else is known about the determinants of VGF trafficking, although a predicted alpha helix loop in its C-terminus may be required for direction into INS1 SGs [149].
A handful of studies have investigated the consequences of granin depletion in β-cells albeit with varying success, possibly due to the method of study. Transient gene silencing seems to outcompete stable knockouts for studying function, and this is probably due to the circumvention of compensatory changes that occur during development. For example, whole body CgB knockout (KO) provided an insulin secretory defect that was unable to be explained aside from a small decrease in the number of docked SGs [150], whereas adenoviral knockdown (KD) of CgB in INS1-832/3 insulinoma cells and isolated mouse islets resulted in marked insulin secretory defects that could be explained by a defect to SG biogenesis [147]. Similarly, islets from whole body CgA KOs actually have enhanced insulin secretion with no defects to SG generation [151], whereas siRNA KD of CgA in the human β-cell line, EndoC-βH1, resulted in reduced basal and glucose-stimulated insulin secretion (GSIS) as well as cellular insulin content [152]. CgA KO mice had compensatory doubling in CgB expression and tripling in SgII expression [151], which may explain the absence of a secretory phenotype in CgA KOs. Islets from whole-body SgIII KO mice have impaired GSIS but only when subject to a high-fat-high-sucrose diet. This is associated with reduced insulin and increased proinsulin content, but there were no reported ultrastructural granule abnormalities [153]. Interestingly, in this study, CgA levels failed to increase when SgIII KO mice were put on diet but did so in the islets of wild-type mice [153]. As discussed previously, SgIII is a known adaptor for CgA and therefore its absence could result in CgA mis-sorting and thus the failure of compensatory upregulation. Finally, VGF depletion has also been assessed via KD of its mRNA in INS1-832/3 cells and a tamoxifen-inducible KO from mouse islets [154]. This study noted reduced GSIS in both models, associated with a loss of total and docked SGs, and a reduction to their size in line with an increased cellular proinsulin-to-insulin ratio and delays to proinsulin conversion [154]. This study concluded that VGF depletion caused a granule replenishment defect, hampering the secretion of newly synthesized granules during the sustained second phase of GSIS [154].
In summary, the granins are critical components of ISG formation, driving the formation of regulated carriers from within the lumen through aggregating and binding to distinct sites of the TGN membrane. Their ubiquity across cells of the neuroendocrine system implies an essential role for SG function, where their combined abundance and aggregative nature may confer unique characteristics to the SG.
References
- Orci, L.; Halban, P.; Amherdt, M.; Ravazzola, M.; Vassalli, J.D.; Perrelet, A. Nonconverted, Amino Acid Analog-Modified Proinsulin Stays in a Golgi-Derived Clathrin-Coated Membrane Compartment. J. Cell Biol. 1984, 99, 2187–2192.
- Rhodes, C.J.; Halban, P.A. Newly Synthesized Proinsulin/insulin and Stored Insulin Are Released from Pancreatic B Cells Predominantly via a Regulated, rather than a Constitutive, Pathway. J. Cell Biol. 1987, 105, 145–153.
- Michael, J.; Carroll, R.; Swift, H.H.; Steiner, D.F. Studies on the Molecular Organization of Rat Insulin Secretory Granules. J. Biol. Chem. 1987, 262, 16531–16535.
- Davidson, H.W.; Rhodes, C.J.; Hutton, J.C. Intraorganellar Calcium and pH Control Proinsulin Cleavage in the Pancreatic Beta Cell via Two Distinct Site-Specific Endopeptidases. Nature 1988, 333, 93–96.
- Smeekens, S.P.; Montag, A.G.; Thomas, G.; Albiges-Rizo, C.; Carroll, R.; Benig, M.; Phillips, L.A.; Martin, S.; Ohagi, S.; Gardner, P. Proinsulin Processing by the Subtilisin-Related Proprotein Convertases Furin, PC2, and PC3. Proc. Natl. Acad. Sci. USA 1992, 89, 8822–8826.
- Orci, L.; Ravazzola, M.; Storch, M.J.; Anderson, R.G.; Vassalli, J.D.; Perrelet, A. Proteolytic Maturation of Insulin Is a Post-Golgi Event Which Occurs in Acidifying Clathrin-Coated Secretory Vesicles. Cell 1987, 49, 865–868.
- Kuliawat, R.; Klumperman, J.; Ludwig, T.; Arvan, P. Differential Sorting of Lysosomal Enzymes out of the Regulated Secretory Pathway in Pancreatic Beta-Cells. J. Cell Biol. 1997, 137, 595–608.
- Baker, E.N.; Blundell, T.L.; Cutfield, J.F.; Dodson, E.J.; Dodson, G.G.; Hodgkin, D.M.C.; Hubbard, R.E.; Isaacs, N.W.; Reynolds, C.D.; Sakabe, K.; et al. The Structure of 2Zn Pig Insulin Crystals at 1.5 Å Resolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1988, 319, 369–456.
- Halban, P.A. Inhibition of Proinsulin to Insulin Conversion in Rat Islets Using Arginine and Lysine Analogs. Lack of Effect on Rate of Release of Modified Products. J. Biol. Chem. 1982, 257, 13177–13180.
- Seaquist, E.R.; Kahn, S.E.; Clark, P.M.; Hales, C.N.; Porte, D., Jr.; Robertson, R.P. Hyperproinsulinemia Is Associated with Increased Beta Cell Demand after Hemipancreatectomy in Humans. J. Clin. Investig. 1996, 97, 455–460.
- Mezza, T.; Ferraro, P.M.; Sun, V.A.; Moffa, S.; Cefalo, C.M.A.; Quero, G.; Cinti, F.; Sorice, G.P.; Pontecorvi, A.; Folli, F.; et al. Increased β-Cell Workload Modulates Proinsulin-to-Insulin Ratio in Humans. Diabetes 2018, 67, 2389–2396.
- Alarcon, C.; Boland, B.B.; Uchizono, Y.; Moore, P.C.; Peterson, B.; Rajan, S.; Rhodes, O.S.; Noske, A.B.; Haataja, L.; Arvan, P.; et al. Pancreatic β-Cell Adaptive Plasticity in Obesity Increases Insulin Production but Adversely Affects Secretory Function. Diabetes 2016, 65, 438–450.
- Ward, W.K.; LaCava, E.C.; Paquette, T.L.; Beard, J.C.; Wallum, B.J.; Porte, D., Jr. Disproportionate Elevation of Immunoreactive Proinsulin in Type 2 (non-Insulin-Dependent) Diabetes Mellitus and in Experimental Insulin Resistance. Diabetologia 1987, 30, 698–702.
- Yoshino, H.; Kawakami, K.; Yoshino, G.; Hirose, T. Age-Related Changes of Proinsulin Processing in Diabetic and Non-Diabetic Japanese Individuals. Geriatr. Gerontol. Int. 2018, 18, 1046–1050.
- Pfützner, A.; Standl, E.; Hohberg, C.; Konrad, T.; Strotmann, H.-J.; Lübben, G.; Langenfeld, M.R.; Schulze, J.; Forst, T. IRIS II Study: Intact Proinsulin Is Confirmed as a Highly Specific Indicator for Insulin Resistance in a Large Cross-Sectional Study Design. Diabetes Technol. Ther. 2005, 7, 478–486.
- Pradhan, A.D.; Manson, J.E.; Meigs, J.B.; Rifai, N.; Buring, J.E.; Liu, S.; Ridker, P.M. Insulin, Proinsulin, Proinsulin:insulin Ratio, and the Risk of Developing Type 2 Diabetes Mellitus in Women. Am. J. Med. 2003, 114, 438–444.
- Nijpels, G.; Popp-Snijders, C.; Kostense, P.J.; Bouter, L.M.; Heine, R.J. Fasting Proinsulin and 2-H Post-Load Glucose Levels Predict the Conversion to NIDDM in Subjects with Impaired Glucose Tolerance: The Hoorn Study. Diabetologia 1996, 39, 113–118.
- Vangipurapu, J.; Stančáková, A.; Kuulasmaa, T.; Kuusisto, J.; Laakso, M. Both Fasting and Glucose-Stimulated Proinsulin Levels Predict Hyperglycemia and Incident Type 2 Diabetes: A Population-Based Study of 9,396 Finnish Men. PLoS ONE 2015, 10, e0124028.
- Yang, Y.; Wang, M.; Tong, J.; Dong, Z.; Deng, M.; Ren, X.; Li, H.; Yang, J.; Meng, Z.; Sun, J.; et al. Impaired Glucose-Stimulated Proinsulin Secretion Is an Early Marker of β-Cell Impairment Before Prediabetes Stage. J. Clin. Endocrinol. Metab. 2019, 104, 4341–4346.
- Porte, D., Jr.; Kahn, S.E. Hyperproinsulinemia and Amyloid in NIDDM. Clues to Etiology of Islet Beta-Cell Dysfunction? Diabetes 1989, 38, 1333–1336.
- Rhodes, C.J.; Alarcón, C. What Beta-Cell Defect Could Lead to Hyperproinsulinemia in NIDDM? Some Clues from Recent Advances Made in Understanding the Proinsulin-Processing Mechanism. Diabetes 1994, 43, 511–517.
- Ohsawa, H.; Kanatsuka, A.; Yamaguchi, T.; Makino, H.; Yoshida, S. Islet Amyloid Polypeptide Inhibits Glucose-Stimulated Insulin Secretion from Isolated Rat Pancreatic Islets. Biochem. Biophys. Res. Commun. 1989, 160, 961–967.
- Natori, S.; Huttner, W.B. Chromogranin B (secretogranin I) Promotes Sorting to the Regulated Secretory Pathway of Processing Intermediates Derived from a Peptide Hormone Precursor. Proc. Natl. Acad. Sci. USA 1996, 93, 4431–4436.
- Carmon, O.; Laguerre, F.; Riachy, L.; Delestre-Delacour, C.; Wang, Q.; Tanguy, E.; Jeandel, L.; Cartier, D.; Thahouly, T.; Haeberlé, A.-M.; et al. Chromogranin A Preferential Interaction with Golgi Phosphatidic Acid Induces Membrane Deformation and Contributes to Secretory Granule Biogenesis. FASEB J. 2020.
- Bartolomucci, A.; Possenti, R.; Mahata, S.K.; Fischer-Colbrie, R.; Loh, Y.P.; Salton, S.R.J. The Extended Granin Family: Structure, Function, and Biomedical Implications. Endocr. Rev. 2011, 32, 755–797.
- Hutton, J.C.; Peshavaria, M. Proton-Translocating Mg2+-Dependent ATPase Activity in Insulin-Secretory Granules. Biochem. J. 1982, 204, 161–170.
- Schoonderwoert, V.T.; Holthuis, J.C.; Tanaka, S.; Tooze, S.A.; Martens, G.J. Inhibition of the Vacuolar H+-ATPase Perturbs the Transport, Sorting, Processing and Release of Regulated Secretory Proteins. Eur. J. Biochem. 2000, 267, 5646–5654.
- Lemaire, K.; Chimienti, F.; Schuit, F. Zinc Transporters and Their Role in the Pancreatic β-Cell. J. Diabetes Investig. 2012, 3, 202–211.
- Hutton, J.C.; Penn, E.J.; Peshavaria, M. Low-Molecular-Weight Constituents of Isolated Insulin-Secretory Granules. Bivalent Cations, Adenine Nucleotides and Inorganic Phosphate. Biochem. J. 1983, 210, 297–305.
- Hur, Y.S.; Yoo, S.H. Distribution Profile of Inositol 1,4,5-Trisphosphate Receptor/Ca2+ Channels in α and β Cells of Pancreas: Dominant Localization in Secretory Granules and Common Error in Identification of Secretory Granule Membranes. Pancreas 2015, 44, 158–165.
- Mitchell, K.J.; Pinton, P.; Varadi, A.; Tacchetti, C.; Ainscow, E.K.; Pozzan, T.; Rizzuto, R.; Rutter, G.A. Dense Core Secretory Vesicles Revealed as a Dynamic Ca(2+) Store in Neuroendocrine Cells with a Vesicle-Associated Membrane Protein Aequorin Chimaera. J. Cell Biol. 2001, 155, 41–51.
- Klumperman, J.; Kuliawat, R.; Griffith, J.M.; Geuze, H.J.; Arvan, P. Mannose 6-Phosphate Receptors Are Sorted from Immature Secretory Granules via Adaptor Protein AP-1, Clathrin, and Syntaxin 6-Positive Vesicles. J. Cell Biol. 1998, 141, 359–371.
- Ericson, L.E.; Håkanson, R.; Lundquist, I. Accumulation of Dopamine in Mouse Pancreatic B-Cells Following Injection of L-DOPA. Localization to Secretory Granules and Inhibition of Insulin Secretion. Diabetologia 1977, 13, 117–124.
- Ekholm, R.; Ericson, L.E.; Lundquist, I. Monoamines in the Pancreatic Islets of the Mouse. Subcellular Localization of 5-Hydroxytryptamine by Electron Microscopic Autoradiography. Diabetologia 1971, 7, 339–348.
- Lundquist, I.; Ekholm, R.; Ericson, L.E. Monoamines in the Pancreatic Islets of the Mouse. 5-Hydroxytryptamine as an Intracellular Modifier of Insulin Secretion, and the Hypoglycaemic Action of Monoamine Oxidase Inhibitors. Diabetologia 1971, 7, 414–422.
- Sakamoto, S.; Miyaji, T.; Hiasa, M.; Ichikawa, R.; Uematsu, A.; Iwatsuki, K.; Shibata, A.; Uneyama, H.; Takayanagi, R.; Yamamoto, A.; et al. Impairment of Vesicular ATP Release Affects Glucose Metabolism and Increases Insulin Sensitivity. Sci. Rep. 2014, 4, 6689.
- Henquin, J.-C. Paracrine and Autocrine Control of Insulin Secretion in Human Islets: Evidence and Pending Questions. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E78–E86.
- Liu, M.; Lara-Lemus, R.; Shan, S.-O.; Wright, J.; Haataja, L.; Barbetti, F.; Guo, H.; Larkin, D.; Arvan, P. Impaired Cleavage of Preproinsulin Signal Peptide Linked to Autosomal-Dominant Diabetes. Diabetes 2012, 61, 828–837.
- Huang, X.F.; Arvan, P. Intracellular Transport of Proinsulin in Pancreatic Beta-Cells. Structural Maturation Probed by Disulfide Accessibility. J. Biol. Chem. 1995, 270, 20417–20423.
- Haataja, L.; Snapp, E.; Wright, J.; Liu, M.; Hardy, A.B.; Wheeler, M.B.; Markwardt, M.L.; Rizzo, M.; Arvan, P. Proinsulin Intermolecular Interactions during Secretory Trafficking in Pancreatic β Cells. J. Biol. Chem. 2013, 288, 1896–1906.
- Frank, B.H.; Veros, A.J. Interaction of Zinc with Proinsulin. Biochem. Biophys. Res. Commun. 1970, 38, 284–289.
- Kuliawat, R.; Arvan, P. Distinct Molecular Mechanisms for Protein Sorting within Immature Secretory Granules of Pancreatic Beta-Cells. J. Cell Biol. 1994, 126, 77–86.
- Kiselar, J.G.; Datt, M.; Chance, M.R.; Weiss, M.A. Structural Analysis of Proinsulin Hexamer Assembly by Hydroxyl Radical Footprinting and Computational Modeling. J. Biol. Chem. 2011, 286, 43710–43716.
- Bailyes, E.M.; Bennett, D.L.; Hutton, J.C. Proprotein-Processing Endopeptidases of the Insulin Secretory Granule. Enzyme 1991, 45, 301–313.
- Davidson, H.W.; Hutton, J.C. The Insulin-Secretory-Granule Carboxypeptidase H. Purification and Demonstration of Involvement in Proinsulin Processing. Biochem. J. 1987, 245, 575–582.
- Weiss, M.; Steiner, D.F.; Philipson, L.H. Insulin Biosynthesis, Secretion, Structure, and Structure-Activity Relationships. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2014.
- Norrman, M.; Schluckebier, G. Crystallographic Characterization of Two Novel Crystal Forms of Human Insulin Induced by Chaotropic Agents and a Shift in pH. BMC Struct. Biol. 2007, 7, 83.
- Steiner, D.F. Cocrystallization of Proinsulin and Insulin. Nature 1973, 243, 528–530.
- Landreh, M.; Alvelius, G.; Willander, H.; Stukenborg, J.-B.; Söder, O.; Johansson, J.; Jörnvall, H. Insulin Solubility Transitions by pH-Dependent Interactions with Proinsulin C-Peptide. FEBS J. 2012, 279, 4589–4597.
- Verchere, C.B.; Paoletta, M.; Neerman-Arbez, M.; Rose, K.; Irminger, J.C.; Gingerich, R.L.; Kahn, S.E.; Halban, P.A. Des-(27–31)C-Peptide. A Novel Secretory Product of the Rat Pancreatic Beta Cell Produced by Truncation of Proinsulin Connecting Peptide in Secretory Granules. J. Biol. Chem. 1996, 271, 27475–27481.
- Gold, G.; Grodsky, G.M. Kinetic Aspects of Compartmental Storage and Secretion of Insulin and Zinc. Experientia 1984, 40, 1105–1114.
- Chan, S.J.; Seino, S.; Gruppuso, P.A.; Schwartz, R.; Steiner, D.F. A Mutation in the B Chain Coding Region Is Associated with Impaired Proinsulin Conversion in a Family with Hyperproinsulinemia. Proc. Natl. Acad. Sci. USA 1987, 84, 2194–2197.
- Carroll, R.J.; Hammer, R.E.; Chan, S.J.; Swift, H.H.; Rubenstein, A.H.; Steiner, D.F. A Mutant Human Proinsulin Is Secreted from Islets of Langerhans in Increased Amounts via an Unregulated Pathway. Proc. Natl. Acad. Sci. USA 1988, 85, 8943–8947.
- Halban, P.A.; Irminger, J.-C. Mutant Proinsulin That Cannot Be Converted Is Secreted Efficiently from Primary Rat Beta-Cells via the Regulated Pathway. Mol. Biol. Cell 2003, 14, 1195–1203.
- Sizonenko, S.; Irminger, J.C.; Buhler, L.; Deng, S.; Morel, P.; Halban, P.A. Kinetics of Proinsulin Conversion in Human Islets. Diabetes 1993, 42, 933–936.
- Sizonenko, S.V.; Halban, P.A. Differential Rates of Conversion of Rat Proinsulins I and II. Evidence for Slow Cleavage at the B-chain/C-Peptide Junction of Proinsulin II. Biochem. J. 1991, 278, 621–625.
- Schechter, I.; Berger, A. On the Size of the Active Site in Proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162.
- Kaufmann, J.E.; Irminger, J.C.; Halban, P.A. Sequence Requirements for Proinsulin Processing at the B-chain/C-Peptide Junction. Biochem. J. 1995, 310 Pt 3, 869–874.
- Halban, P.A. Proinsulin Processing in the Regulated and the Constitutive Secretory Pathway. Diabetologia 1994, 37 (Suppl. 2), S65–S72.
- Rhodes, C.J.; Lincoln, B.; Shoelson, S.E. Preferential Cleavage of Des-31,32-Proinsulin over Intact Proinsulin by the Insulin Secretory Granule Type II Endopeptidase. Implication of a Favored Route for Prohormone Processing. J. Biol. Chem. 1992, 267, 22719–22727.
- Kaufmann, J.E.; Irminger, J.C.; Mungall, J.; Halban, P.A. Proinsulin Conversion in GH3 Cells after Coexpression of Human Proinsulin with the Endoproteases PC2 And/or PC3. Diabetes 1997, 46, 978–982.
- Irminger, J.C.; Vollenweider, F.M.; Neerman-Arbez, M.; Halban, P.A. Human Proinsulin Conversion in the Regulated and the Constitutive Pathways of Transfected AtT20 Cells. J. Biol. Chem. 1994, 269, 1756–1762.
- Neerman-Arbez, M.; Sizonenko, S.V.; Halban, P.A. Slow Cleavage at the Proinsulin B-Chain/connecting Peptide Junction Associated with Low Levels of Endoprotease PC1/3 in Transformed Beta Cells. J. Biol. Chem. 1993, 268, 16098–16100.
- Neerman-Arbez, M.; Cirulli, V.; Halban, P.A. Levels of the Conversion Endoproteases PC1 (PC3) and PC2 Distinguish between Insulin-Producing Pancreatic Islet Beta Cells and Non-Beta Cells. Biochem. J. 1994, 300 Pt 1, 57–61.
- Zhu, X.; Orci, L.; Carroll, R.; Norrbom, C.; Ravazzola, M.; Steiner, D.F. Severe Block in Processing of Proinsulin to Insulin Accompanied by Elevation of Des-64,65 Proinsulin Intermediates in Islets of Mice Lacking Prohormone Convertase 1/3. Proc. Natl. Acad. Sci. USA 2002, 99, 10299–10304.
- Furuta, M.; Carroll, R.; Martin, S.; Swift, H.H.; Ravazzola, M.; Orci, L.; Steiner, D.F. Incomplete Processing of Proinsulin to Insulin Accompanied by Elevation of Des-31,32 Proinsulin Intermediates in Islets of Mice Lacking Active PC2. J. Biol. Chem. 1998, 273, 3431–3437.
- Ramzy, A.; Asadi, A.; Kieffer, T.J. Revisiting Proinsulin Processing: Evidence That Human β-Cells Process Proinsulin with Prohormone Convertase (PC) 1/3 but Not PC2. Diabetes 2020, 69, 1451–1462.
- Irminger, J.C.; Meyer, K.; Halban, P. Proinsulin Processing in the Rat Insulinoma Cell Line INS after Overexpression of the Endoproteases PC2 or PC3 by Recombinant Adenovirus. Biochem. J. 1996, 320 Pt 1, 11–15.
- Shennan, K.I.; Taylor, N.A.; Jermany, J.L.; Matthews, G.; Docherty, K. Differences in pH Optima and Calcium Requirements for Maturation of the Prohormone Convertases PC2 and PC3 Indicates Different Intracellular Locations for These Events. J. Biol. Chem. 1995, 270, 1402–1407.
- Muller, L.; Zhu, X.; Lindberg, I. Mechanism of the Facilitation of PC2 Maturation by 7B2: Involvement in ProPC2 Transport and Activation but Not Folding. J. Cell Biol. 1997, 139, 625–638.
- Zhu, X.; Rouille, Y.; Lamango, N.S.; Steiner, D.F.; Lindberg, I. Internal Cleavage of the Inhibitory 7B2 Carboxyl-Terminal Peptide by PC2: A Potential Mechanism for Its Inactivation. Proc. Natl. Acad. Sci. USA 1996, 93, 4919–4924.
- Hwang, J.R.; Lindberg, I. Inactivation of the 7B2 Inhibitory CT Peptide Depends on a Functional Furin Cleavage Site. J. Neurochem. 2001, 79, 437–444.
- Lamango, N.S.; Apletalina, E.; Liu, J.; Lindberg, I. The Proteolytic Maturation of Prohormone Convertase 2 (PC2) Is a pH-Driven Process. Arch. Biochem. Biophys. 1999, 362, 275–282.
- Ostrega, D.; Polonsky, K.; Nagi, D.; Yudkin, J.; Cox, L.J.; Clark, P.M.; Hales, C.N. Measurement of Proinsulin and Intermediates. Validation of Immunoassay Methods by High-Performance Liquid Chromatography. Diabetes 1995, 44, 437–440.
- Clark, P.M.; Levy, J.C.; Cox, L.; Burnett, M.; Turner, R.C.; Hales, C.N. Immunoradiometric Assay of Insulin, Intact Proinsulin and 32–33 Split Proinsulin and Radioimmunoassay of Insulin in Diet-Treated Type 2 (non-Insulin-Dependent) Diabetic Subjects. Diabetologia 1992, 35, 469–474.
- Lukinius, A.; Wilander, E.; Westermark, G.T.; Engström, U.; Westermark, P. Co-Localization of Islet Amyloid Polypeptide and Insulin in the B Cell Secretory Granules of the Human Pancreatic Islets. Diabetologia 1989, 32, 240–244.
- Nakazato, M.; Miyazato, M.; Asai, J.; Mitsukawa, T.; Kangawa, K.; Matsuo, H.; Matsukura, S. Islet Amyloid Polypeptide, a Novel Pancreatic Peptide, Is a Circulating Hormone Secreted under Glucose Stimulation. Biochem. Biophys. Res. Commun. 1990, 169, 713–718.
- Stridsberg, M.; Sandler, S.; Wilander, E. Cosecretion of Islet Amyloid Polypeptide (IAPP) and Insulin from Isolated Rat Pancreatic Islets Following Stimulation or Inhibition of Beta-Cell Function. Regul. Pept. 1993, 45, 363–370.
- Lutz, T.A. The Role of Amylin in the Control of Energy Homeostasis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1475–R1484.
- Hull, R.L.; Westermark, G.T.; Westermark, P.; Kahn, S.E. Islet Amyloid: A Critical Entity in the Pathogenesis of Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 3629–3643.
- Westermark, P.; Grimelius, L. The Pancreatic Islet Cells in Insular Amyloidosis in Human Diabetic and Non-Diabetic Adults. Acta Pathol. Microbiol. Scand. A 1973, 81, 291–300.
- Westermark, P.; Wilander, E. The Influence of Amyloid Deposits on the Islet Volume in Maturity Onset Diabetes Mellitus. Diabetologia 1978, 15, 417–421.
- Zhao, H.-L.; Lai, F.M.M.; Tong, P.C.Y.; Zhong, D.-R.; Yang, D.; Tomlinson, B.; Chan, J.C.N. Prevalence and Clinicopathological Characteristics of Islet Amyloid in Chinese Patients with Type 2 Diabetes. Diabetes 2003, 52, 2759–2766.
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet Amyloid Polypeptide, Islet Amyloid, and Diabetes Mellitus. Physiol. Rev. 2011, 91, 795–826.
- Haataja, L.; Gurlo, T.; Huang, C.J.; Butler, P.C. Islet Amyloid in Type 2 Diabetes, and the Toxic Oligomer Hypothesis. Endocr. Rev. 2008, 29, 303–316.
- Raleigh, D.; Zhang, X.; Hastoy, B.; Clark, A. The β-Cell Assassin: IAPP Cytotoxicity. J. Mol. Endocrinol. 2017, 59, R121–R140.
- Wang, J.; Xu, J.; Finnerty, J.; Furuta, M.; Steiner, D.F.; Verchere, C.B. The Prohormone Convertase Enzyme 2 (PC2) Is Essential for Processing pro-Islet Amyloid Polypeptide at the NH2-Terminal Cleavage Site. Diabetes 2001, 50, 534–539.
- Marzban, L.; Soukhatcheva, G.; Verchere, C.B. Role of Carboxypeptidase E in Processing of Pro-Islet Amyloid Polypeptide in β-Cells. Endocrinology 2005, 146, 1808–1817.
- Marzban, L.; Trigo-Gonzalez, G.; Verchere, C.B. Processing of pro-Islet Amyloid Polypeptide in the Constitutive and Regulated Secretory Pathways of Beta Cells. Mol. Endocrinol. 2005, 19, 2154–2163.
- Marzban, L.; Trigo-Gonzalez, G.; Zhu, X.; Rhodes, C.J.; Halban, P.A.; Steiner, D.F.; Verchere, C.B. Role of Beta-Cell Prohormone Convertase (PC)1/3 in Processing of pro-Islet Amyloid Polypeptide. Diabetes 2004, 53, 141–148.
- Chen, Y.-C.; Taylor, A.J.; Verchere, C.B. Islet Prohormone Processing in Health and Disease. Diabetes Obes. Metab. 2018, 20 (Suppl. 2), 64–76.
- Rulifson, I.C.; Cao, P.; Miao, L.; Kopecky, D.; Huang, L.; White, R.D.; Samayoa, K.; Gardner, J.; Wu, X.; Chen, K.; et al. Identification of Human Islet Amyloid Polypeptide as a BACE2 Substrate. PLoS ONE 2016, 11, e0147254.
- Courtade, J.A.; Wang, E.Y.; Yen, P.; Dai, D.L.; Soukhatcheva, G.; Orban, P.C.; Verchere, C.B. Loss of Prohormone Convertase 2 Promotes Beta Cell Dysfunction in a Rodent Transplant Model Expressing Human pro-Islet Amyloid Polypeptide. Diabetologia 2017, 60, 453–463.
- Westermark, P.; Engström, U.; Johnson, K.H.; Westermark, G.T.; Betsholtz, C. Islet Amyloid Polypeptide: Pinpointing Amino Acid Residues Linked to Amyloid Fibril Formation. Proc. Natl. Acad. Sci. USA 1990, 87, 5036–5040.
- Yonemoto, I.T.; Kroon, G.J.A.; Dyson, H.J.; Balch, W.E.; Kelly, J.W. Amylin Proprotein Processing Generates Progressively More Amyloidogenic Peptides That Initially Sample the Helical State. Biochemistry 2008, 47, 9900–9910.
- Jaikaran, E.T.A.S.; Nilsson, M.R.; Clark, A. Pancreatic Beta-Cell Granule Peptides Form Heteromolecular Complexes Which Inhibit Islet Amyloid Polypeptide Fibril Formation. Biochem. J. 2004, 377, 709–716.
- Westermark, P.; Li, Z.C.; Westermark, G.T.; Leckström, A.; Steiner, D.F. Effects of Beta Cell Granule Components on Human Islet Amyloid Polypeptide Fibril Formation. FEBS Lett. 1996, 379, 203–206.
- Brender, J.R.; Hartman, K.; Nanga, R.P.R.; Popovych, N.; de la Salud Bea, R.; Vivekanandan, S.; Marsh, E.N.G.; Ramamoorthy, A. Role of Zinc in Human Islet Amyloid Polypeptide Aggregation. J. Am. Chem. Soc. 2010, 132, 8973–8983.
- Khemtémourian, L.; Doménech, E.; Doux, J.P.F.; Koorengevel, M.C.; Killian, J.A. Low pH Acts as Inhibitor of Membrane Damage Induced by Human Islet Amyloid Polypeptide. J. Am. Chem. Soc. 2011, 133, 15598–15604.
- Janciauskiene, S.; Eriksson, S.; Carlemalm, E.; Ahrén, B. B Cell Granule Peptides Affect Human Islet Amyloid Polypeptide (IAPP) Fibril Formation in Vitro. Biochem. Biophys. Res. Commun. 1997, 236, 580–585.
- Kudva, Y.C.; Mueske, C.; Butler, P.C.; Eberhardt, N.L. A Novel Assay in Vitro of Human Islet Amyloid Polypeptide Amyloidogenesis and Effects of Insulin Secretory Vesicle Peptides on Amyloid Formation. Biochem. J. 1998, 331 Pt 3, 809–813.
- Johnson, K.H.; O’Brien, T.D.; Jordan, K.; Westermark, P. Impaired Glucose Tolerance Is Associated with Increased Islet Amyloid Polypeptide (IAPP) Immunoreactivity in Pancreatic Beta Cells. Am. J. Pathol. 1989, 135, 245–250.
- Gurlo, T.; Ryazantsev, S.; Huang, C.-J.; Yeh, M.W.; Reber, H.A.; Hines, O.J.; O’Brien, T.D.; Glabe, C.G.; Butler, P.C. Evidence for Proteotoxicity in Beta Cells in Type 2 Diabetes: Toxic Islet Amyloid Polypeptide Oligomers Form Intracellularly in the Secretory Pathway. Am. J. Pathol. 2010, 176, 861–869.
- Lin, C.-Y.; Gurlo, T.; Kayed, R.; Butler, A.E.; Haataja, L.; Glabe, C.G.; Butler, P.C. Toxic Human Islet Amyloid Polypeptide (h-IAPP) Oligomers Are Intracellular, and Vaccination to Induce Anti-Toxic Oligomer Antibodies Does Not Prevent H-IAPP-Induced Beta-Cell Apoptosis in H-IAPP Transgenic Mice. Diabetes 2007, 56, 1324–1332.
- Yagui, K.; Yamaguchi, T.; Kanatsuka, A.; Shimada, F.; Huang, C.I.; Tokuyama, Y.; Ohsawa, H.; Yamamura, K.; Miyazaki, J.; Mikata, A. Formation of Islet Amyloid Fibrils in Beta-Secretory Granules of Transgenic Mice Expressing Human Islet Amyloid Polypeptide/amylin. Eur. J. Endocrinol. 1995, 132, 487–496.
- Wang, H.; Raleigh, D.P. The Ability of Insulin to Inhibit the Formation of Amyloid by pro-Islet Amyloid Polypeptide Processing Intermediates Is Significantly Reduced in the Presence of Sulfated Glycosaminoglycans. Biochemistry 2014, 53, 2605–2614.
- Janson, J.; Ashley, R.H.; Harrison, D.; McIntyre, S.; Butler, P.C. The Mechanism of Islet Amyloid Polypeptide Toxicity Is Membrane Disruption by Intermediate-Sized Toxic Amyloid Particles. Diabetes 1999, 48, 491–498.
- Brender, J.R.; Lee, E.L.; Cavitt, M.A.; Gafni, A.; Steel, D.G.; Ramamoorthy, A. Amyloid Fiber Formation and Membrane Disruption Are Separate Processes Localized in Two Distinct Regions of IAPP, the Type-2-Diabetes-Related Peptide. J. Am. Chem. Soc. 2008, 130, 6424–6429.
- Brender, J.R.; Salamekh, S.; Ramamoorthy, A. Membrane Disruption and Early Events in the Aggregation of the Diabetes Related Peptide IAPP from a Molecular Perspective. Acc. Chem. Res. 2012, 45, 454–462.
- Westermark, P.; Engström, U.; Westermark, G.T.; Johnson, K.H.; Permerth, J.; Betsholtz, C. Islet Amyloid Polypeptide (IAPP) and pro-IAPP Immunoreactivity in Human Islets of Langerhans. Diabetes Res. Clin. Pract. 1989, 7, 219–226.
- Westermark, G.T.; Steiner, D.F.; Gebre-Medhin, S.; Engström, U.; Westermark, P. Pro Islet Amyloid Polypeptide (ProIAPP) Immunoreactivity in the Islets of Langerhans. Ups. J. Med. Sci. 2000, 105, 97–106.
- Zheng, X.; Ren, W.; Zhang, S.; Liu, J.; Li, S.; Li, J.; Yang, P.; He, J.; Su, S.; Li, P. Serum Levels of Proamylin and Amylin in Normal Subjects and Patients with Impaired Glucose Regulation and Type 2 Diabetes Mellitus. Acta Diabetol. 2010, 47, 265–270.
- Xu, J.; Wijesekara, N.; Regeenes, R.; Rijjal, D.A.; Piro, A.L.; Song, Y.; Wu, A.; Bhattacharjee, A.; Liu, Y.; Marzban, L.; et al. Pancreatic β Cell-Selective Zinc Transporter 8 Insufficiency Accelerates Diabetes Associated with Islet Amyloidosis. JCI Insight 2021, 6.
- Elias, S.; Delestre, C.; Ory, S.; Marais, S.; Courel, M.; Vazquez-Martinez, R.; Bernard, S.; Coquet, L.; Malagon, M.M.; Driouich, A.; et al. Chromogranin A Induces the Biogenesis of Granules with Calcium- and Actin-Dependent Dynamics and Exocytosis in Constitutively Secreting Cells. Endocrinology 2012, 153, 4444–4456.
- Montero-Hadjadje, M.; Elias, S.; Chevalier, L.; Benard, M.; Tanguy, Y.; Turquier, V.; Galas, L.; Yon, L.; Malagon, M.M.; Driouich, A.; et al. Chromogranin A Promotes Peptide Hormone Sorting to Mobile Granules in Constitutively and Regulated Secreting Cells: Role of Conserved N- and C-Terminal Peptides. J. Biol. Chem. 2009, 284, 12420–12431.
- Inomoto, C.; Umemura, S.; Egashira, N.; Minematsu, T.; Takekoshi, S.; Itoh, Y.; Itoh, J.; Taupenot, L.; O’Connor, D.T.; Osamura, R.Y. Granulogenesis in Non-Neuroendocrine COS-7 Cells Induced by EGFP-Tagged Chromogranin A Gene Transfection: Identical and Distinct Distribution of CgA and EGFP. J. Histochem. Cytochem. 2007, 55, 487–493.
- Huh, Y.H.; Jeon, S.H.; Yoo, S.H. Chromogranin B-Induced Secretory Granule Biogenesis: Comparison with the Similar Role of Chromogranin A. J. Biol. Chem. 2003, 278, 40581–40589.
- Chanat, E.; Huttner, W.B. Milieu-Induced, Selective Aggregation of Regulated Secretory Proteins in the Trans-Golgi Network. J. Cell Biol. 1991, 115, 1505–1519.
- Yoo, S.H.; Albanesi, J.P. High Capacity, Low Affinity Ca2+ Binding of Chromogranin A. Relationship between the pH-Induced Conformational Change and Ca2+ Binding Property. J. Biol. Chem. 1991, 266, 7740–7745.
- Sun-Wada, G.-H.; Toyomura, T.; Murata, Y.; Yamamoto, A.; Futai, M.; Wada, Y. The a3 Isoform of V-ATPase Regulates Insulin Secretion from Pancreatic Beta-Cells. J. Cell Sci. 2006, 119 Pt 21, 4531–4540.
- Taupenot, L.; Harper, K.L.; O’Connor, D.T. Role of H+-ATPase-Mediated Acidification in Sorting and Release of the Regulated Secretory Protein Chromogranin A: Evidence for a Vesiculogenic Function. J. Biol. Chem. 2005, 280, 3885–3897.
- Lissandron, V.; Podini, P.; Pizzo, P.; Pozzan, T. Unique Characteristics of Ca2+ Homeostasis of the Trans-Golgi Compartment. Proc. Natl. Acad. Sci. USA 2010, 107, 9198–9203.
- Arvan, P.; Halban, P.A. Sorting Ourselves out: Seeking Consensus on Trafficking in the Beta-Cell. Traffic 2004, 5, 53–61.
- Klemm, R.W.; Ejsing, C.S.; Surma, M.A.; Kaiser, H.-J.; Gerl, M.J.; Sampaio, J.L.; de Robillard, Q.; Ferguson, C.; Proszynski, T.J.; Shevchenko, A.; et al. Segregation of Sphingolipids and Sterols during Formation of Secretory Vesicles at the Trans-Golgi Network. J. Cell Biol. 2009, 185, 601–612.
- Orci, L.; Montesano, R.; Meda, P.; Malaisse-Lagae, F.; Brown, D.; Perrelet, A.; Vassalli, P. Heterogeneous Distribution of Filipin--Cholesterol Complexes across the Cisternae of the Golgi Apparatus. Proc. Natl. Acad. Sci. USA 1981, 78, 293–297.
- von Blume, J.; Hausser, A. Lipid-Dependent Coupling of Secretory Cargo Sorting and Trafficking at the Trans-Golgi Network. FEBS Lett. 2019, 593, 2412–2427.
- Wang, Y.; Thiele, C.; Huttner, W.B. Cholesterol Is Required for the Formation of Regulated and Constitutive Secretory Vesicles from the Trans-Golgi Network. Traffic 2000, 1, 952–962.
- Wang, T.Y.; Silvius, J.R. Different Sphingolipids Show Differential Partitioning into Sphingolipid/cholesterol-Rich Domains in Lipid Bilayers. Biophys. J. 2000, 79, 1478–1489.
- Kreutzberger, A.J.B.; Kiessling, V.; Doyle, C.A.; Schenk, N.; Upchurch, C.M.; Elmer-Dixon, M.; Ward, A.E.; Preobraschenski, J.; Hussein, S.S.; Tomaka, W.; et al. Distinct Insulin Granule Subpopulations Implicated in the Secretory Pathology of Diabetes Types 1 and 2. eLife 2020, 9.
- Hussain, S.S.; Harris, M.T.; Kreutzberger, A.J.B.; Inouye, C.M.; Doyle, C.A.; Castle, A.M.; Arvan, P.; Castle, J.D. Control of Insulin Granule Formation and Function by the ABC Transporters ABCG1 and ABCA1 and by Oxysterol Binding Protein OSBP. Mol. Biol. Cell 2018, 29, 1238–1257.
- Tsuchiya, M.; Hosaka, M.; Moriguchi, T.; Zhang, S.; Suda, M.; Yokota-Hashimoto, H.; Shinozuka, K.; Takeuchi, T. Cholesterol Biosynthesis Pathway Intermediates and Inhibitors Regulate Glucose-Stimulated Insulin Secretion and Secretory Granule Formation in Pancreatic Beta-Cells. Endocrinology 2010, 151, 4705–4716.
- Bogan, J.S.; Xu, Y.; Hao, M. Cholesterol Accumulation Increases Insulin Granule Size and Impairs Membrane Trafficking. Traffic 2012, 13, 1466–1480.
- Payet, L.-A.; Pineau, L.; Snyder, E.C.R.; Colas, J.; Moussa, A.; Vannier, B.; Bigay, J.; Clarhaut, J.; Becq, F.; Berjeaud, J.-M.; et al. Saturated Fatty Acids Alter the Late Secretory Pathway by Modulating Membrane Properties. Traffic 2013, 14, 1228–1241.
- Yoo, S.H.; Oh, Y.S.; Kang, M.K.; Huh, Y.H.; So, S.H.; Park, H.S.; Park, H.Y. Localization of Three Types of the Inositol 1,4,5-Trisphosphate Receptor/Ca2+ Channel in the Secretory Granules and Coupling with the Ca2+ Storage Proteins Chromogranins A and B*. J. Biol. Chem. 2001, 276, 45806–45812.
- Yoo, S.H.; Lewis, M.S. Thermodynamic Study of the pH-Dependent Interaction of Chromogranin A with an Intraluminal Loop Peptide of the Inositol 1,4,5-Trisphosphate Receptor. Biochemistry 1995, 34, 632–638.
- Hosaka, M.; Suda, M.; Sakai, Y.; Izumi, T.; Watanabe, T.; Takeuchi, T. Secretogranin III Binds to Cholesterol in the Secretory Granule Membrane as an Adapter for Chromogranin A. J. Biol. Chem. 2004, 279, 3627–3634.
- Hosaka, M.; Watanabe, T.; Sakai, Y.; Uchiyama, Y.; Takeuchi, T. Identification of a Chromogranin A Domain That Mediates Binding to Secretogranin III and Targeting to Secretory Granules in Pituitary Cells and Pancreatic Beta-Cells. Mol. Biol. Cell 2002, 13, 3388–3399.
- Han, L.; Suda, M.; Tsuzuki, K.; Wang, R.; Ohe, Y.; Hirai, H.; Watanabe, T.; Takeuchi, T.; Hosaka, M. A Large Form of Secretogranin III Functions as a Sorting Receptor for Chromogranin a Aggregates in PC12 Cells. Mol. Endocrinol. 2008, 22, 1935–1949.
- Courel, M.; Vasquez, M.S.; Hook, V.Y.; Mahata, S.K.; Taupenot, L. Sorting of the Neuroendocrine Secretory Protein Secretogranin II into the Regulated Secretory Pathway: Role of N- and C-Terminal Alpha-Helical Domains. J. Biol. Chem. 2008, 283, 11807–11822.
- Sun, M.; Watanabe, T.; Bochimoto, H.; Sakai, Y.; Torii, S.; Takeuchi, T.; Hosaka, M. Multiple Sorting Systems for Secretory Granules Ensure the Regulated Secretion of Peptide Hormones. Traffic 2013, 14, 205–218.
- Hotta, K.; Hosaka, M.; Tanabe, A.; Takeuchi, T. Secretogranin II Binds to Secretogranin III and Forms Secretory Granules with Orexin, Neuropeptide Y, and POMC. J. Endocrinol. 2009, 202, 111–121.
- Courel, M.; Soler-Jover, A.; Rodriguez-Flores, J.L.; Mahata, S.K.; Elias, S.; Montero-Hadjadje, M.; Anouar, Y.; Giuly, R.J.; O’Connor, D.T.; Taupenot, L. Pro-Hormone Secretogranin II Regulates Dense Core Secretory Granule Biogenesis in Catecholaminergic Cells. J. Biol. Chem. 2010, 285, 10030–10043.
- Glombik, M.M.; Krömer, A.; Salm, T.; Huttner, W.B.; Gerdes, H.H. The Disulfide-Bonded Loop of Chromogranin B Mediates Membrane Binding and Directs Sorting from the Trans-Golgi Network to Secretory Granules. EMBO J. 1999, 18, 1059–1070.
- Pimplikar, S.W.; Huttner, W.B. Chromogranin B (secretogranin I), a Secretory Protein of the Regulated Pathway, Is Also Present in a Tightly Membrane-Associated Form in PC12 Cells. J. Biol. Chem. 1992, 267, 4110–4118.
- Giordano, T.; Brigatti, C.; Podini, P.; Bonifacio, E.; Meldolesi, J.; Malosio, M.L. Beta Cell Chromogranin B Is Partially Segregated in Distinct Granules and Can Be Released Separately from Insulin in Response to Stimulation. Diabetologia 2008, 51, 997–1007.
- Yoo, S.H.; Chu, S.Y.; Kim, K.D.; Huh, Y.H. Presence of Secretogranin II and High-Capacity, Low-Affinity Ca2+ Storage Role in Nucleoplasmic Ca2+ Store Vesicles. Biochemistry 2007, 46, 14663–14671.
- Bearrows, S.C.; Bauchle, C.J.; Becker, M.; Haldeman, J.M.; Swaminathan, S.; Stephens, S.B. Chromogranin B Regulates Early-Stage Insulin Granule Trafficking from the Golgi in Pancreatic Islet β-Cells. J. Cell Sci. 2019, 132.
- Yoo, S.H.; Lewis, M.S. Effects of pH and Ca2+ on Heterodimer and Heterotetramer Formation by Chromogranin A and Chromogranin B. J. Biol. Chem. 1996, 271, 17041–17046.
- Garcia, A.L.; Han, S.-K.; Janssen, W.G.; Khaing, Z.Z.; Ito, T.; Glucksman, M.J.; Benson, D.L.; Salton, S.R.J. A Prohormone Convertase Cleavage Site within a Predicted α-Helix Mediates Sorting of the Neuronal and Endocrine Polypeptide VGF into the Regulated Secretory Pathway *. J. Biol. Chem. 2005, 280, 41595–41608.
- Obermüller, S.; Calegari, F.; King, A.; Lindqvist, A.; Lundquist, I.; Salehi, A.; Francolini, M.; Rosa, P.; Rorsman, P.; Huttner, W.B.; et al. Defective Secretion of Islet Hormones in Chromogranin-B Deficient Mice. PLoS ONE 2010, 5, e8936.
- Wollam, J.; Mahata, S.; Riopel, M.; Hernandez-Carretero, A.; Biswas, A.; Bandyopadhyay, G.K.; Chi, N.-W.; Eiden, L.E.; Mahapatra, N.R.; Corti, A.; et al. Chromogranin A Regulates Vesicle Storage and Mitochondrial Dynamics to Influence Insulin Secretion. Cell Tissue Res. 2017, 368, 487–501.
- Thomsen, S.K.; Raimondo, A.; Hastoy, B.; Sengupta, S.; Dai, X.-Q.; Bautista, A.; Censin, J.; Payne, A.J.; Umapathysivam, M.M.; Spigelman, A.F.; et al. Type 2 Diabetes Risk Alleles in PAM Impact Insulin Release from Human Pancreatic β-Cells. Nat. Genet. 2018, 50, 1122–1131.
- Maeda, Y.; Kudo, S.; Tsushima, K.; Sato, E.; Kubota, C.; Kayamori, A.; Bochimoto, H.; Koga, D.; Torii, S.; Gomi, H.; et al. Impaired Processing of Prohormones in Secretogranin III-Null Mice Causes Maladaptation to an Inadequate Diet and Stress. Endocrinology 2018, 159, 1213–1227.
- Stephens, S.B.; Edwards, R.J.; Sadahiro, M.; Lin, W.-J.; Jiang, C.; Salton, S.R.; Newgard, C.B. The Prohormone VGF Regulates β Cell Function via Insulin Secretory Granule Biogenesis. Cell Rep. 2017, 20, 2480–2489.


