 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pradip Devhare | + 2688 word(s) | 2688 | 2021-09-03 13:08:47 | | | |
| 2 | Beatrix Zheng | + 437 word(s) | 3125 | 2021-09-10 12:31:12 | | |
Video Upload Options
Hepatitis E virus (HEV) is one of the major causes of acute, self-limiting viral hepatitis (AVH) which is now recognized as a global health problem in both developing and industrialized regions. According to WHO estimates in 2015, annually 20 million HEV infections cause 3.3 million symptomatic cases and account for 3.3% of the mortality due to viral hepatitis. The feco-oral route transmission of HEV through contaminated drinking water is attributed to large-scale epidemics in developing countries while zoonotic transmission through the consumption of undercooked meat, blood transfusions, and organ transplantation are considered as major sources of HEV infections in developed nations.
1. Introduction
Hepatitis E virus (HEV) is one of the major causes of acute, self-limiting viral hepatitis (AVH) which is now recognized as a global health problem in both developing and industrialized regions [1][2][3]. According to WHO estimates in 2015, annually 20 million HEV infections cause 3.3 million symptomatic cases and account for 3.3% of the mortality due to viral hepatitis [4]. The feco-oral route transmission of HEV through contaminated drinking water is attributed to large-scale epidemics in developing countries while zoonotic transmission through the consumption of undercooked meat, blood transfusions, and organ transplantation are considered as major sources of HEV infections in developed nations [3]. Though HEV infection causes self-limiting disease in most young adults, pregnant women from developing countries are at high risk accounting for ~30% of the fatality rate in the third trimester [5][6]. The mechanisms underlying HEV pathogenesis during pregnancy are undermined and related mostly to hormonal imbalance and immunological alterations [5][6]. Chronic hepatitis E has been reported in immunocompromised patients such as organ transplant recipients, HIV-infected individuals, and patients with hematological malignancies (reviewed in [7]). Moreover, extrahepatic manifestations like neuronal and renal diseases and pancreatitis are also reported during the course of HEV infection [8][9].
HEV is a non-enveloped, spherical particle with a diameter of 27–34 nm when it is isolated from bile and feces whereas it also exists as a quasi-enveloped virus (~40 nm) in blood and cell culture supernatants [3]. The single-stranded, positive-sense RNA genome of ~7.2 Kb encompasses three open reading frames (ORFs): ORF1, ORF2, and ORF3 which are flanked by short untranslated regions (UTRs) at 5′ and 3′-ends. The 5′-end is m7G-capped (7-methylguanosine) and the 3′- end is polyadenylated. Recently, an additional ORF (ORF4) has been described to be expressed by genotype 1 HEV which aids in efficient virus replication under stress conditions [10] ( Figure 1 ). Yadav et al. (2021) demonstrated that the ectopic expression of genotype 1 HEV ORF4 enhanced replication of genotype 3 which does not code for ORF4 [11]. Nonstructural proteins such as methyltransferase (MT), a papain-like cysteine protease (PCP), a helicase, and an RNA-dependent RNA polymerase (RdRp) are encoded by ORF1. Less characterized domains including Y-domain, the hypervariable region (HVR), and X-domain (Macrodomain) also constitute ORF1 [12]. ORF2 encodes for viral capsid protein while ORF3 which almost entirely overlaps with ORF2 encodes for a small multifunctional ion channel protein involved in viral egress from infected cells [13]. Notably, HEV replication and its life cycle remained poorly understood in terms of ORF1 polyprotein processing, the role of PCP, transcriptional regulation of subgenomic RNA encoding ORF2 and ORF3, and components of HEV replicase complex including essential host factors [3][14][15][16]. Cell culture adapted HEV strains and the development of genome-length and subgenomic infectious complementary DNA (cDNA) clones by reverse genetics [17][18][19][20] have paved the way to understand certain aspects of host–virus interactions during HEV infection [21].
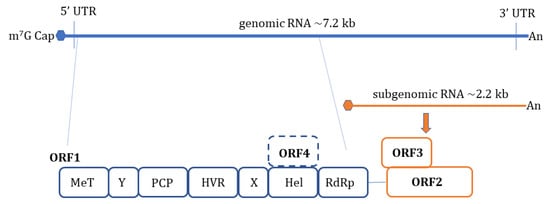
It is proposed that HEV enters host cells through capsid protein interaction with cellular receptors, Heparin Sulfate Proteoglycans (HSPGs), asialoglycoprotein receptor 1/2 (ASGPR1/2) , integrin α3 (ITGA3), ATP synthase subunit 5β (ATP5B), glucose-regulated protein 78 (GRP78), and Heat shock cognate protein 70 (HSC70) [14][21]. After entry and uncoating, the ORF1 is translated by host ribosomes into polyprotein containing RdRp. The RdRp uses (+) -sense genomic RNA as a template and makes full-length (−)-ve sense RNA which serves as a template for making genomic (+) -sense RNA and small subgenomic RNA encoding ORF2 and ORF3. Also, multiple steps in virus assembly are poorly understood. ORF2 binding to viral genomic RNA, multimerization of ORF2 as well as an association of ORF3 to ORF2 during assembly and viral egress has been proposed (reviewed in [14][15][16]). During these replication events, viral genomic RNA, double-stranded replication intermediates (dsRNA), and proteins are under strict surveillance by the host cell PRRs to induce innate defense ( Figure 2 ).
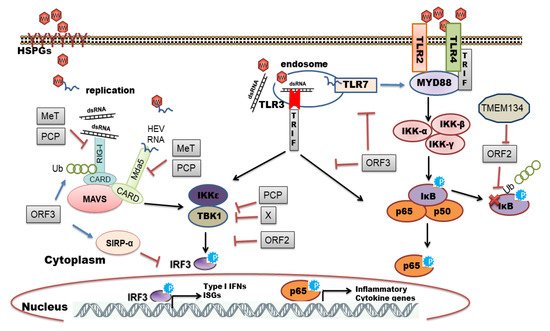
The host organism can employ different families of PRRs to sense and immediately respond to a diverse range of pathogens. Individual members of the PRR families can be distinguished by ligand specificity, their cellular localization, and the activation of unique but converging downstream signaling pathways [22][23]. The sensing of PAMPs by PRRs upregulates the transcription of genes involved in inflammatory responses [24]. These genes encode proinflammatory cytokines, type I interferons (IFNs), chemokines, antimicrobial proteins, and proteins involved in the modulation of PRR signaling. In the context of viral infections, Toll-like receptors (TLRs) and retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) play a pivotal role in recognizing viral PAMPs and eliciting antiviral response [25][26][27]. In this review, we have summarized an interplay between HEV and host cell PRRs along with the mechanisms of subversion of innate immune signaling by HEV.
2. HEV Sensing by RLRs
HEV ORF3 protein is known to enhance poly I:C (double-stranded RNA analog) mediated IFN induction by interaction with RIG-I and extending its half-life through K63-linked ubiquitination [28]. Interestingly, authors noted that ORF3 from genotype 1 and III boosted RIG-I signaling whereas ORF3 from genotype II and IV had a minimal effect [28]. It will be interesting to explore genotype-specific virulent factors in the host tropism of HEV which remains an open question. Using the HEV genotype 1 replicon system, we could show altered replication efficiency of HEV in different human hepatoma cell lines (Huh7, Huh7.5, and HepG2/C3A) due to cell-type-specific innate immune responses [29]. We identified the role of RIG-I and TLR3 in sensing HEV RNA and activation of downstream interferon regulatory factor 3 (IRF3) mediated antiviral responses. Inhibition of this signaling cascade downstream of PRRs by pharmacological inhibitor BX795 significantly improved HEV replication efficiency [29]. Furthermore, overexpression of interferon regulatory factor 1 (IRF1) effectively inhibited HEV replication through the JAK-STAT signaling cascade independent of interferons. The authors revealed transcriptional activation of STAT1 promoter by IRF1 resulted in the elevation of intracellular STAT1 levels and transcription of ISGs [30]. The same research group in 2017 identified RIG-1 as a key anti-HEV ISG in cell culture models by overexpression studies [31]. The pharmacological activation of RIG-I by its natural ligand (5′-pppRNA) was identified to combat HEV infection which was independent of IRF3/IRF7 activation and IFN induction [31]. The authors proposed two distinct mechanisms of RIG- I mediated ISG induction viz. JAK-STAT dependent and independent [31]. The integrity of JAK-STAT signaling in antiviral defense against HEV was confirmed by immunohistochemical staining of transcriptionally active phospho-STAT1 (Y701) in liver biopsy samples of acute or chronic HEV [32]. Single-stranded HEV genomic RNA potently induced innate immune response in cell lines as well as in primary three-dimensional liver organoid cultures [32]. Moreover, HEV genotype 3 specific stable replicon system in human (Huh-7-S10-3) or hamster (BHK-21) origin cells also revealed the role of RIG-I and IRF3 in antiviral defense during HEV replication [33].
The second RLR, Mda5 was seen to be upregulated at 48 h post-infection in epithelial cells (A549) during HEV infection in our TaqMan low-density array (TLDA) based transcriptome analysis study [34]. In hepatoma cell lines HepG2/C3A and Huh7:S10-3 transfected with HEV subgenomic RNA , Mda5 expression was higher after 24 h post-transfection and it was associated with viral RNA replication as Mda5 expression was comparatively less in cells transfected with replication-deficient RNA [29]. Recently, Li et al. (2020) demonstrated the role of Mda5 in inhibiting HEV replication in both HEV infectious and subgenomic replicon models [35]. The overexpression of Mda5 mediated its antiviral action by inducing a wide range of antiviral ISGs independent of interferon production through partial activation of JAK-STAT signaling [35].
The functional knockdown of RLR signaling mediators; RIG-I, Mda5, and MAVS in liver cells revealed the reduction of HEV induced type III IFNs which implicated the role of both RLRs in sensing HEV RNA [36]. Moreover, transcriptome analysis of primary human hepatocytes infected with genotype 3 HEV revealed expression of PRRs; RIG-I, Mda5, TLR3, and downstream signaling molecules MyD88 and MAVS, supporting the physiological role of innate immune signaling during HEV infection [37]. Both RIG-I and Mda5 are known to recognize specific molecular patterns in foreign RNA. 5′-ppp RNA [38], capped dsRNA, as well as size and composition of the RNA ligand are demonstrated as some important features specifically identified by RIG-I and Mda5 [39][40]. Homopolyuridine or homopolyriboadenine motifs present in the genomes of RNA viruses are the chief features of RIG-I recognition in human and murine cells [41]. In this regard, Sooryanarain et al. (2020) attempted to identify HEV RNA-associated molecular patterns in eliciting RIG- I mediated antiviral response [42]. The authors identified the U-rich region in the 3′ untranslated region (UTR) of the HEV genome as a potent RIG-I agonist.
3. HEV Infection-Mediated Interferons (IFNs)
IFNs are grouped into type I, type II, and type III IFNs based on their amino acid composition. Type I IFNs (often abbreviated as IFN-α/β) , Type II IFNs (IFN-γ), and Type III IFNs comprise IFNλ1 (IL-29), -λ2 (IL-28A), -λ3 (IL-28B) and a newly discovered subtype IFNλ4. These cytokines use the same pathway of activation as that of IFN-α/β in response to direct viral infection by engaging distinct receptor complexes [25][43]. Exogenous treatment by all three types of IFNs exhibited antiviral activity against HEV in subgenomic and full-length genome cell culture models [44]. IFN-α has been identified as a potent antiviral factor inhibiting HEV replication [29][44][45]. Genotype 3 HEV infection in HepG2 cells and primary human hepatocytes induced type III IFNs and virus replication persisted in presence of type III IFNs. Interestingly, among type III IFNs, the expression of IFN-λ1 and IFN-λ2/3 was found to be upregulated while IFN-λ4 remained unchanged following HEV infection [36]. This distinct regulation of IFN-λ subtypes during HEV pathogenesis remains obscured. IFN-λ4 is a newly discovered subtype present upstream of the IFN-λ3 gene which was reported to be associated with treatment-induced clearance of Hepatitis C virus (HCV) [46]. Similarly, an association of favorable IFN-λ4 polymorphism and treatment with high-flux hemodialysis were reported to be determinants of anti-HEV IgG positivity (spontaneous HEV resolution) among hemodialysis patients exposed to HEV [47]. Stem cell-derived hepatocyte-like cells were shown to be permissive for infection by all four HEV genotypes (genotype 1 to 4) [48]. Cell culture adapted genotype 3 HEV infection elicited induction of IFN-β, -λ1, and -λ3 expression at mRNA level whereas IFN-α expression remained undetectable in these hepatocyte-like cells. However, at protein level only type III IFN levels were higher at 7- and 9-days post-infection while type I IFNs remained undetected in cell culture supernatants [48]. Consistent with these in vitro results, the pig model system of genotype 3 HEV infection also showed predominant induction of type III IFNs in liver tissues [42]. This HEV-induced IFN induction was observed to be cell type-dependent manner as there was predominantly type III IFN response in human and pig hepatocytes, while type I IFN induction was noted in HEV infected pig enterocytes [42]. HEV can replicate in primary human intestinal cells and infection of these cells with clinically isolated (stool-derived) genotype 1 and -3 HEV profoundly induced more type III IFNs than type I IFNs [49]. Recently, high levels of IFN-λ3 were detected in serum samples of acute hepatitis E patients during the early phase of the infection which was also correlated with viral RNA in serum. Furthermore, the antiviral activity of IFN-λ3 in a dose-dependent manner was verified in the cell culture model [50]. Altogether, the majority of the studies reveal the role of type III IFNs followed by type I IFNs in controlling HEV infection.
4. Induction of Interferon Stimulated Genes (ISGs) by HEV
The IFN signaling cascade begins with the engagement of heterodimeric receptors (IFNAR1/IFNAR2 or IL-28Rα/IL-10R2) by IFNs which activates Janus kinases (JAK), Jak1, and Tyk2, which in turn phosphorylate STAT1 (signal transducers and activators of transcription1) and STAT2 transcription factors. Subsequently, STAT1 and -2 heterodimerize and interact with IRF9 to form the heterotrimeric cytoplasmic complex, called ISGF3. This complex translocate into the nucleus, binds to genes containing IFN-stimulated response elements (ISREs), and induces expression of ISGs which are major antiviral effectors [25][43] ( Figure 3 ).
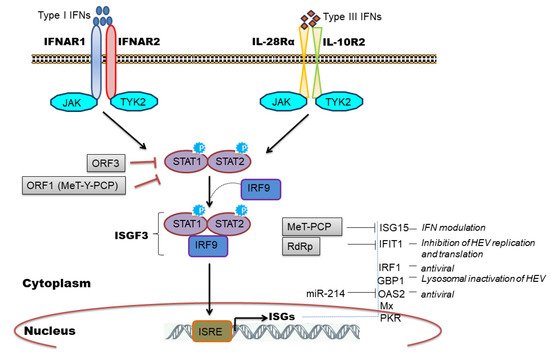
We and others have observed induction of ISGs during HEV replication [34][29][31][32][33][36][37][51][52][53] and clinically ISG induction has been related to HEV persistence in chronic HEV patients [54]. Furthermore, the constitutive expression of ISGs was mediated by unphosphorylated ISGF3 complex to inhibit HEV replication [55] and the knockdown of individual ISGF3 components; IRF9, STAT1, and STAT2 reversed the antiviral effect of ISGs and increased viral replication [45][55]. Few studies have attempted to characterize the antiviral role of individual ISGs in regulating HEV infection. To illustrate, ectopic expression of 25 ISGs in genotype 3 HEV cell culture model identified RIG-I, Mda5, and IRF1 as major ISGs inhibiting HEV replication [31]. IRF1 is another important ISG that was shown to inhibit HEV replication by the induction of antiviral ISGs through the JAK-STAT pathway but independent of IFN induction [30]. Interestingly, targeting the nucleotide synthesis pathway was linked with ISG induction and anti-HEV effects, independent of JAK-STAT signaling [56]. However, the mechanistic insights related to nucleotide biosynthesis pathway and cellular immune responses remain elusive. ISG15 is a ubiquitin-like modifier protein induced by IFNs and gets conjugated onto different target proteins (ISGylation) or secreted as a cytokine to regulate diverse functions [57]. An increase in ISG15 mRNA levels in liver biopsies of HEV infected chimpanzee [51], chronic HEV patients [54], and cell culture models of HEV infection [34][29] implicated its role during HEV replication. In this context, an immunomodulatory role of ISG15 was proposed [58][59] wherein ISG15 expression was induced at mRNA and protein level in HEV infected cells as well as in pig livers [59]. Knockdown of ISG15 enhanced expression of type I IFNs [58] and IFN mediated antiviral response against HEV by inducing expression of other ISGs like PKR and OAS1 [59]. ISG15 is also induced by unphosphorylated ISGF3 complex [55] and might be causing IFN unresponsiveness in HEV infected cells by maintaining a sustained expression of ubiquitin-specific protease 18 (USP18), a negative regulator of IFN signaling [60]. IFIT1 (Interferon-induced protein with tetratricopeptide repeat 1) is an IFN induced antiviral protein known to recognize viral RNAs without the 2′-OH-methylated cap (Cap0) or with a free 5′-ppp RNA and thereby regulating viral RNA translation [61]. We observed an upregulation of this gene (ISG56) in genotype 1 HEV infected or replicon transfected cells [34][29] while genotype 3 HEV also induced IFIT1 expression in HepG2/C3A cells [36]. Recently, Pingale et al. (2019) demonstrated the functional association of IFIT1 with HEV replication [62]. The authors showed that overexpression of IFIT1 inhibited HEV replication by interacting with HEV RNA and affecting its translation [62]. Transcriptionally GBP1 (guanylate-binding protein 1) was found to be elevated at 48-h post-infection in HEV infected cells (A549) compared to UV inactivated HEV infection as well as in replicon transfected hepatocyte cell lines [34][29]. Very recently, the role of this antiviral factor against HEV was established. Authors revealed that GBP1 exerted its antiviral role by the homodimerization and independent of GTPase activity wherein GBP1 targeted virus to the lysosomal compartment to inactivate it [63].
Few in vivo studies have documented the use of animal models to understand HEV pathogenesis [64]. Yu et al. (2010) compared Microarray-based transcriptome profiling between HCV- and HEV-infected chimpanzees. Both the viruses elicited different magnitude of immune response whilst in HCV it was more robust compared to HEV infection. However, there was a significant increase in ISG expression in the livers of HEV-infected chimpanzees [51]. Genotype specific difference in host immune response of rhesus macaques was also noted when they were infected with genotype 1 and 3 HEV [52]. Notably, the gene expressions varied in different phases of infection and during early viremia, hepatic immune-response related genes were down-regulated in genotype 1 infected animals compared to genotype 3 infection [52]. Homologous HEV genotype 1 re-infection in seroconverted rhesus macaques showed upregulation of mitogen-activated protein kinase (MAPK) signaling suggesting antiviral oxidative stress response [65]. The human liver chimeric mouse model developed for HEV genotype 1 and 3 revealed the induction of ISGs in genotype 1 infected liver tissue and the induction of innate immune response was more pronounced at 9 days post-infection compared to 4-week post-infection [53]. Pigs are also considered as a model system for the propagation of genotype 3 and 4 HEV infection [66][67]. Recently, a predominant type III IFN response was observed in liver tissues of HEV genotype 3 infected pigs [42].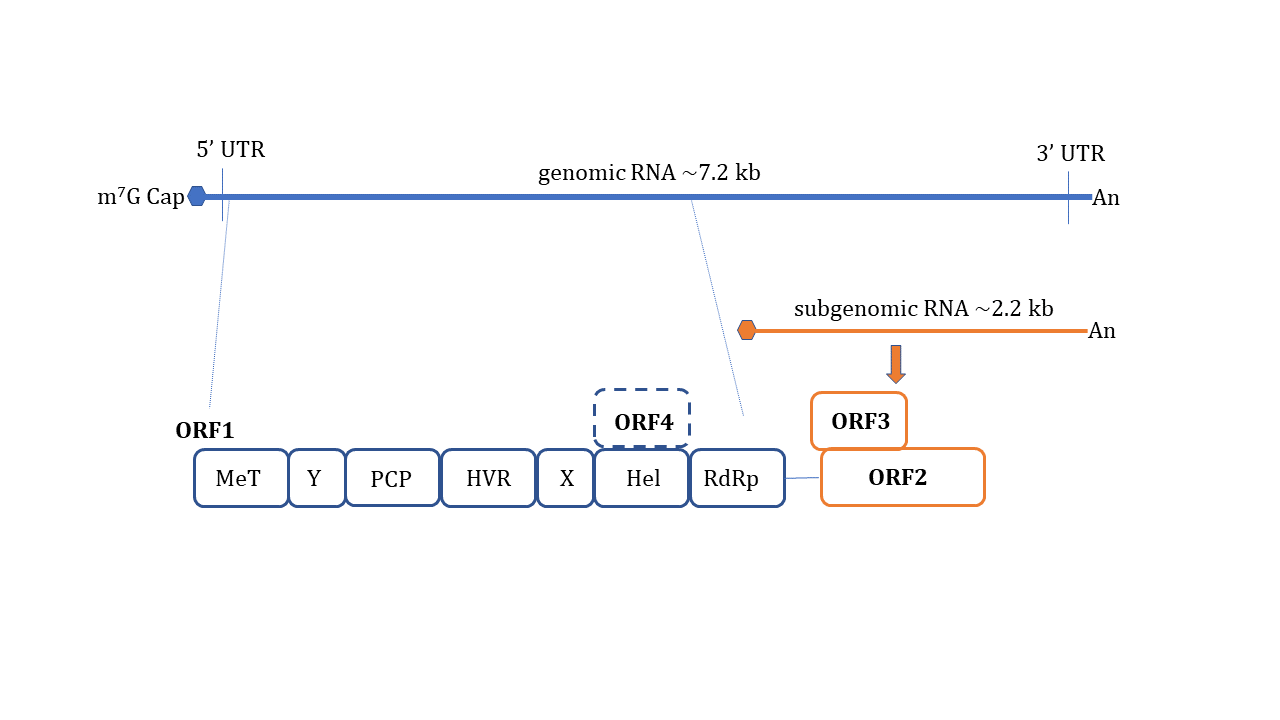
References
- Aggarwal, R.; Jameel, S. Hepatitis E. Hepatology 2011, 54, 2218–2226.
- Kumar, S.; Subhadra, S.; Singh, B.; Panda, B.K. Hepatitis E virus: The current scenario. Int. J. Infect. Dis. 2013, 17, e228–e233.
- Nimgaonkar, I.; Ding, Q.; Schwartz, R.E.; Ploss, A. Hepatitis E virus: Advances and challenges. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 96–110.
- World Health Organization; Hepatitis, E. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-e (accessed on 7 July 2021).
- Navaneethan, U.; Al Mohajer, M.; Shata, M.T. Hepatitis E and pregnancy: Understanding the pathogenesis. Liver Int. 2008, 28, 1190–1199.
- Perez-Gracia, M.T.; Suay-Garcia, B.; Mateos-Lindemann, M.L. Hepatitis E and pregnancy: Current state. Rev. Med. Virol. 2017, 27, e1929.
- Thakur, V.; Ratho, R.K.; Kumar, S.; Saxena, S.K.; Bora, I.; Thakur, P. Viral Hepatitis E and Chronicity: A Growing Public Health Concern. Front. Microbiol. 2020, 11, 577339.
- Bazerbachi, F.; Haffar, S.; Garg, S.K.; Lake, J.R. Extra-hepatic manifestations associated with hepatitis E virus infection: A comprehensive review of the literature. Gastroenterol. Rep. 2016, 4, 1–15.
- Pischke, S.; Hartl, J.; Pas, S.D.; Lohse, A.W.; Jacobs, B.C.; Van der Eijk, A.A. Hepatitis E virus: Infection beyond the liver? J. Hepatol. 2017, 66, 1082–1095.
- Nair, V.P.; Anang, S.; Subramani, C.; Madhvi, A.; Bakshi, K.; Srivastava, A.; Ranjith Kumar, C.T.; Surjit, M. Endoplasmic reticulum stress induced synthesis of a novel viral factor mediates efficient replication of genotype-1 hepatitis E virus. PLoS Pathog. 2016, 12, e1005521.
- Yadav, K.K.; Boley, P.A.; Fritts, Z.; Kenney, S.P. Ectopic expression of genotype 1 hepatitis E virus ORF4 increases genotype 3 HEV viral replication in cell culture. Viruses 2021, 13, 75.
- Koonin, E.V.; Gorbalenya, A.E.; Purdy, M.A.; Rozanov, M.N.; Reyes, G.R.; Bradley, D.W. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: Delineation of an additional group of positive-strand RNA plant and animal viruses. Proc. Natl. Acad. Sci. USA 1992, 89, 8259–8263.
- Ding, Q.; Heller, B.; Capuccino, J.M.; Song, B.; Nimgaonkar, I.; Hrebikova, G.; Contreras, J.E.; Ploss, A. Hepatitis E virus ORF3 is a functional ion channel required for release of infectious particles. Proc. Natl. Acad. Sci. USA 2017, 114, 1147–1152.
- Himmelsbach, K.; Bender, D.; Hildt, E. Life cycle and morphogenesis of the hepatitis E virus. Emerg. Microbes. Infect. 2018, 7, 196.
- Kenney, S.P.; Meng, X.J. Hepatitis E virus genome structure and replication strategy. Cold Spring Harb. Perspect. Med. 2019, 9, a031724.
- LeDesma, R.; Nimgaonkar, I.; Ploss, A. Hepatitis E virus replication. Viruses 2019, 11, 719.
- Tanaka, T.; Takahashi, M.; Kusano, E.; Okamoto, H. Development and evaluation of an efficient cell-culture system for Hepatitis E virus. J. Gen. Virol. 2007, 88, 903–911.
- Tanaka, T.; Takahashi, M.; Takahashi, H.; Ichiyama, K.; Hoshino, Y.; Nagashima, S.; Mizuo, H.; Okamoto, H. Development and characterization of a genotype 4 hepatitis E virus cell culture system using a HE-JF5/15F strain recovered from a fulminant hepatitis patient. J. Clin. Microbiol. 2009, 47, 1906–1910.
- Emerson, S.U.; Zhang, M.; Meng, X.J.; Nguyen, H.; St Claire, M.; Govindarajan, S.; Huang, Y.K.; Purcell, R.H. Recombinant hepatitis E virus genomes infectious for primates: Importance of capping and discovery of a cis-reactive element. Proc. Natl. Acad. Sci. USA 2001, 98, 15270–15275.
- Shukla, P.; Nguyen, H.T.; Faulk, K.; Mather, K.; Torian, U.; Engle, R.E.; Emerson, S.U. Adaptation of a genotype 3 hepatitis E virus to efficient growth in cell culture depends on an inserted human gene segment acquired by recombination. J. Virol. 2012, 86, 5697–5707.
- Wißing, M.H.; Brüggemann, Y.; Steinmann, E.; Todt, D. Virus-host cell interplay during hepatitis E virus infection. Trends Microbiol. 2021, 29, 309–319.
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801.
- Ishii, K.J.; Koyama, S.; Nakagawa, A.; Coban, C.; Akira, S. Host innate immune receptors and beyond: Making sense of microbial infections. Cell Host Microbe 2008, 3, 352–363.
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820.
- Randall, R.E.; Goodbourn, S. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47.
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940.
- Jensen, S.; Thomsen, A.R. Sensing of RNA viruses: A review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 2012, 86, 2900–2910.
- Nan, Y.; Ma, Z.; Wang, R.; Yu, Y.; Kannan, H.; Fredericksen, B.; Zhang, Y.J. Enhancement of interferon induction by ORF3 product of hepatitis E virus. J. Virol. 2014, 88, 8696–8705.
- Devhare, P.B.; Desai, S.; Lole, K.S. Innate immune responses in human hepatocyte-derived cell lines alter genotype 1 hepatitis E virus replication efficiencies. Sci. Rep. 2016, 6, 26827.
- Xu, L.; Zhou, X.; Wang, W.; Wang, Y.; Yin, Y.; Laan, L.J.; Sprengers, D.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. IFN regulatory factor 1 restricts hepatitis E virus replication by activating STAT1 to induce antiviral IFN-stimulated genes. FASEB J. 2016, 30, 3352–3367.
- Xu, L.; Wang, W.; Li, Y.; Zhou, X.; Yin, Y.; Wang, Y.; de Man, R.A.; van der Laan, L.J.W.; Huang, F.; Kamar, N.; et al. RIG-I is a key antiviral interferon-stimulated gene against hepatitis E virus regardless of interferon production. Hepatology 2017, 65, 1823–1839.
- Wang, W.; Wang, Y.; Qu, C.; Wang, S.; Zhou, J.; Cao, W.; Xu, L.; Ma, B.; Hakim, M.S.; Yin, Y.; et al. The RNA genome of hepatitis E virus robustly triggers an antiviral interferon response. Hepatology 2018, 67, 2096–2112.
- Xu, L.D.; Zhang, F.; Peng, L.; Luo, W.T.; Chen, C.; Xu, P.; Huang, Y.W. Stable expression of a hepatitis E Virus (HEV) RNA replicon in two mammalian cell lines to assess mechanism of innate immunity and antiviral response. Front. Microbiol. 2020, 11, 603699.
- Devhare, P.B.; Chatterjee, S.N.; Arankalle, V.A.; Lole, K.S. Analysis of antiviral response in human epithelial cells infected with hepatitis E virus. PLoS ONE 2013, 8, e63793.
- Li, Y.; Yu, P.; Qu, C.; Li, P.; Li, Y.; Ma, Z.; Wang, W.; de Man, R.A.; Peppelenbosch, M.P.; Pan, Q. MDA5 against enteric viruses through induction of interferon-like response partially via the JAK-STAT cascade. Antiviral. Res. 2020, 176, 104743.
- Yin, X.; Li, X.; Ambardekar, C.; Hu, Z.; Lhomme, S.; Feng, Z. Hepatitis E virus persists in the presence of a type III interferon response. PLoS Pathog. 2017, 13, e1006417.
- Todt, D.; Friesland, M.; Moeller, N.; Praditya, D.; Kinast, V.; Brüggemann, Y.; Knegendorf, L.; Burkard, T.; Steinmann, J.; Burm, R.; et al. Robust hepatitis E virus infection and transcriptional response in human hepatocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 1731–1741.
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Näslund, T.I.; Liljeström, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 2006, 314, 997–1001.
- Saito, T.; Gale, M., Jr. Differential recognition of double-stranded RNA by RIG-I-like receptors in antiviral immunity. J. Exp. Med. 2008, 205, 1523–1527.
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610.
- Saito, T.; Owen, D.M.; Jiang, F.; Marcotrigiano, J.; Gale, M., Jr. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 2008, 454, 523–527.
- Sooryanarain, H.; Heffron, C.L.; Meng, X.J. The U-rich untranslated region of the hepatitis e virus induces differential type I and Type III interferon responses in a host cell-dependent manner. Mbio 2020, 11, e03103-19.
- Zhou, J.; Wang, Y.; Chang, Q.; Ma, P.; Hu, Y.; Cao, X. Type III Interferons in Viral Infection and Antiviral Immunity. Cell. Physiol. Biochem. 2018, 51, 173–185.
- Todt, D.; François, C.; Anggakusuma; Behrendt, P.; Engelmann, M.; Knegendorf, L.; Vieyres, G.; Wedemeyer, H.; Hartmann, R.; Pietschmann, T.; et al. Antiviral activities of different interferon types and subtypes against hepatitis E virus replication. Antimicrob. Agents Chemother. 2016, 60, 2132–2139.
- Zhou, X.; Xu, L.; Wang, W.; Watashi, K.; Wang, Y.; Sprengers, D.; de Ruiter, P.E.; van der Laan, L.J.; Metselaar, H.J.; Kamar, N.; et al. Disparity of basal and therapeutically activated interferon signalling in constraining hepatitis E virus infection. J. Viral. Hepat. 2016, 23, 294–304.
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013, 45, 164–171.
- Grzegorzewska, A.E.; Świderska, M.K.; Niepolski, L.; Bura, M.; Mostowska, A.; Łagiedo-Żelazowska, M.; Jagodziński, P.P. Interferon-λ4 gene polymorphisms, circulating interferon λ3, and clinical variables in hemodialysis patients exposed to hepatitis E virus. Pol. Arch. Intern. Med. 2018, 128, 344–353.
- Wu, X.; Thi, V.L.D.; Liu, P.; Takacs, C.N.; Xiang, K.; Andrus, L.; Gouttenoire, J.; Moradpour, D.; Rice, C.M. Pan-genotype hepatitis E virus replication in stem cell-derived hepatocellular systems. Gastroenterology 2018, 154, 663–674.
- Marion, O.; Lhomme, S.; Nayrac, M.; Dubois, M.; Pucelle, M.; Requena, M.; Migueres, M.; Abravanel, F.; Peron, J.M.; Carrere, N.; et al. Hepatitis E virus replication in human intestinal cells. Gut 2020, 69, 901–910.
- Murata, K.; Kang, J.H.; Nagashima, S.; Matsui, T.; Karino, Y.; Yamamoto, Y.; Atarashi, T.; Oohara, M.; Uebayashi, M.; Sakata, H.; et al. IFN-λ3 as a host immune response in acute hepatitis E virus infection. Cytokine 2020, 125, 154816.
- Yu, C.; Boon, D.; McDonald, S.L.; Myers, T.G.; Tomioka, K.; Nguyen, H.; Engle, R.E.; Govindarajan, S.; Emerson, S.U.; Purcell, R.H. Pathogenesis of hepatitis E virus and hepatitis C virus in chimpanzees: Similarities and differences. J. Virol. 2010, 84, 11264–11278.
- Choi, Y.H.; Zhang, X.; Tran, C.; Skinner, B. Expression profiles of host immune response-related genes against HEV genotype 3 and genotype 1 infections in rhesus macaques. J. Viral. Hepat. 2018, 25, 986–995.
- Sayed, I.M.; Verhoye, L.; Cocquerel, L.; Abravanel, F.; Foquet, L.; Montpellier, C.; Debing, Y.; Farhoudi, A.; Wychowski, C.; Dubuisson, J.; et al. Study of hepatitis E virus infection of genotype 1 and 3 in mice with humanised liver. Gut 2017, 66, 920–929.
- Moal, V.; Textoris, J.; Ben Amara, A.; Mehraj, V.; Berland, Y.; Colson, P.; Mege, J.L. Chronic hepatitis E virus infection is specifically associated with an interferon-related transcriptional program. J. Infect. Dis. 2013, 207, 125–132.
- Wang, W.; Yin, Y.; Xu, L.; Su, J.; Huang, F.; Wang, Y.; Boor, P.P.C.; Chen, K.; Wang, W.; Cao, W.; et al. Unphosphorylated ISGF3 drives constitutive expression of interferon-stimulated genes to protect against viral infections. Sci. Signal. 2017, 10.
- Wang, Y.; Wang, W.; Xu, L.; Zhou, X.; Shokrollahi, E.; Felczak, K.; van der Laan, L.J.; Pankiewicz, K.W.; Sprengers, D.; Raat, N.J.; et al. Cross talk between nucleotide synthesis pathways with cellular immunity in constraining hepatitis E virus replication. Antimicrob. Agents Chemother. 2016, 60, 2834–2848.
- Perng, Y.C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018, 16, 423–439.
- Wang, M.; Huang, Y.; He, M.; Peng, W.J.; Tian, D.Y. Effects of hepatitis E virus infection on interferon production via ISG15. World J. Gastroenterol. 2018, 24, 2173–2180.
- Sooryanarain, H.; Rogers, A.J.; Cao, D.; Haac, M.E.R.; Karpe, Y.A.; Meng, X.J. ISG15 modulates type I interferon signaling and the antiviral response during hepatitis E virus replication. J. Virol. 2017, 91, e00621-17.
- Sung, P.S.; Cheon, H.; Cho, C.H.; Hong, S.H.; Park, D.Y.; Seo, H.I.; Park, S.H.; Yoon, S.K.; Stark, G.R.; Shin, E.C. Roles of unphosphorylated ISGF3 in HCV infection and interferon responsiveness. Proc. Natl. Acad. Sci. USA 2015, 112, 10443–10448.
- Fensterl, V.; Sen, G.C. Interferon-induced Ifit proteins: Their role in viral pathogenesis. J. Virol. 2015, 89, 2462–2468.
- Pingale, K.D.; Kanade, G.D.; Karpe, Y.A. Hepatitis E virus polymerase binds to IFIT1 to protect the viral RNA from IFIT1-mediated translation inhibition. J. Gen. Virol. 2019, 100, 471–483.
- Glitscher, M.; Himmelsbach, K.; Woytinek, K.; Schollmeier, A.; Johne, R.; Praefcke, G.J.K.; Hildt, E. Identification of the interferon-inducible GTPase GBP1 as major restriction factor for the Hepatitis E virus. J. Virol. 2021, 95, e01564-20.
- Sayed, I.M.; Elkhawaga, A.A.; El-Mokhtar, M.A. In vivo models for studying Hepatitis E virus infection; Updates and applications. Virus Res. 2019, 274, 197765.
- Choi, Y.H.; Zhang, X.; Srinivasamoorthy, G.; Purdy, M.A. Transcriptome analysis in rhesus macaques infected with hepatitis E virus genotype 1/3 infections and genotype 1 re-infection. PLoS ONE 2020, 15, e0237618.
- Sanford, B.J.; Dryman, B.A.; Huang, Y.W.; Feagins, A.R.; Leroith, T.; Meng, X.J. Prior infection of pigs with a genotype 3 swine hepatitis E virus (HEV) protects against subsequent challenges with homologous and heterologous genotypes 3 and 4 human HEV. Virus Res. 2011, 159, 17–22.
- Feagins, A.R.; Opriessnig, T.; Huang, Y.W.; Halbur, P.G.; Meng, X.J. Cross-species infection of specific-pathogen-free pigs by a genotype 4 strain of human hepatitis E virus. J. Med. Virol. 2008, 80, 1379–1386.



