 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Li Chuin Chong | + 4039 word(s) | 4039 | 2021-08-23 06:00:54 | | | |
| 2 | Conner Chen | Meta information modification | 4039 | 2021-09-22 03:40:56 | | | | |
| 3 | Conner Chen | Meta information modification | 4039 | 2021-10-09 11:17:02 | | |
Video Upload Options
Spinal muscular atrophy (SMA), one of the leading inherited causes of child mortality, is a rare neuromuscular disease arising from loss-of-function mutations of the survival motor neuron 1 (SMN1) gene, which encodes the SMN protein. When lacking the SMN protein in neurons, patients suffer from muscle weakness and atrophy, and in the severe cases, respiratory failure and death. Several therapeutic approaches show promise with human testing and three medications have been approved by the U.S. Food and Drug Administration (FDA) to date. Despite the shown promise of these approved therapies, there are some crucial limitations, one of the most important being the cost. The FDA-approved drugs are high-priced and are shortlisted among the most expensive treatments in the world. The price is still far beyond affordable and may serve as a burden for patients. The blooming of the biomedical data and advancement of computational approaches have opened new possibilities for SMA therapeutic development.
1. Introduction
2. Spinal Muscular Atrophy (SMA)
2.1. Disease Etiology
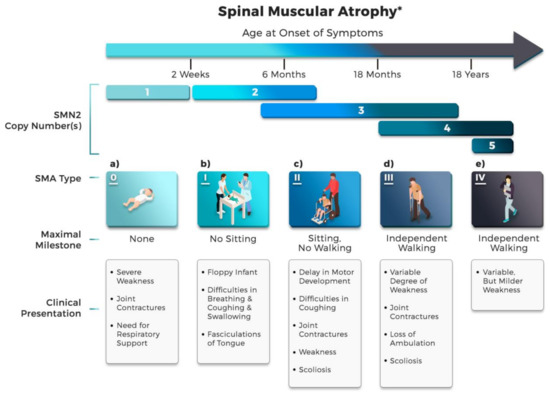
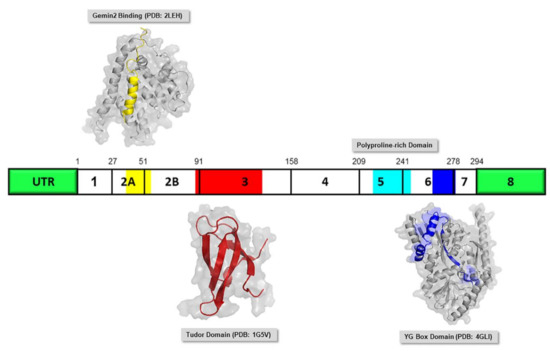
2.2. SMN Protein
3. Current Drug of SMA - Early Success
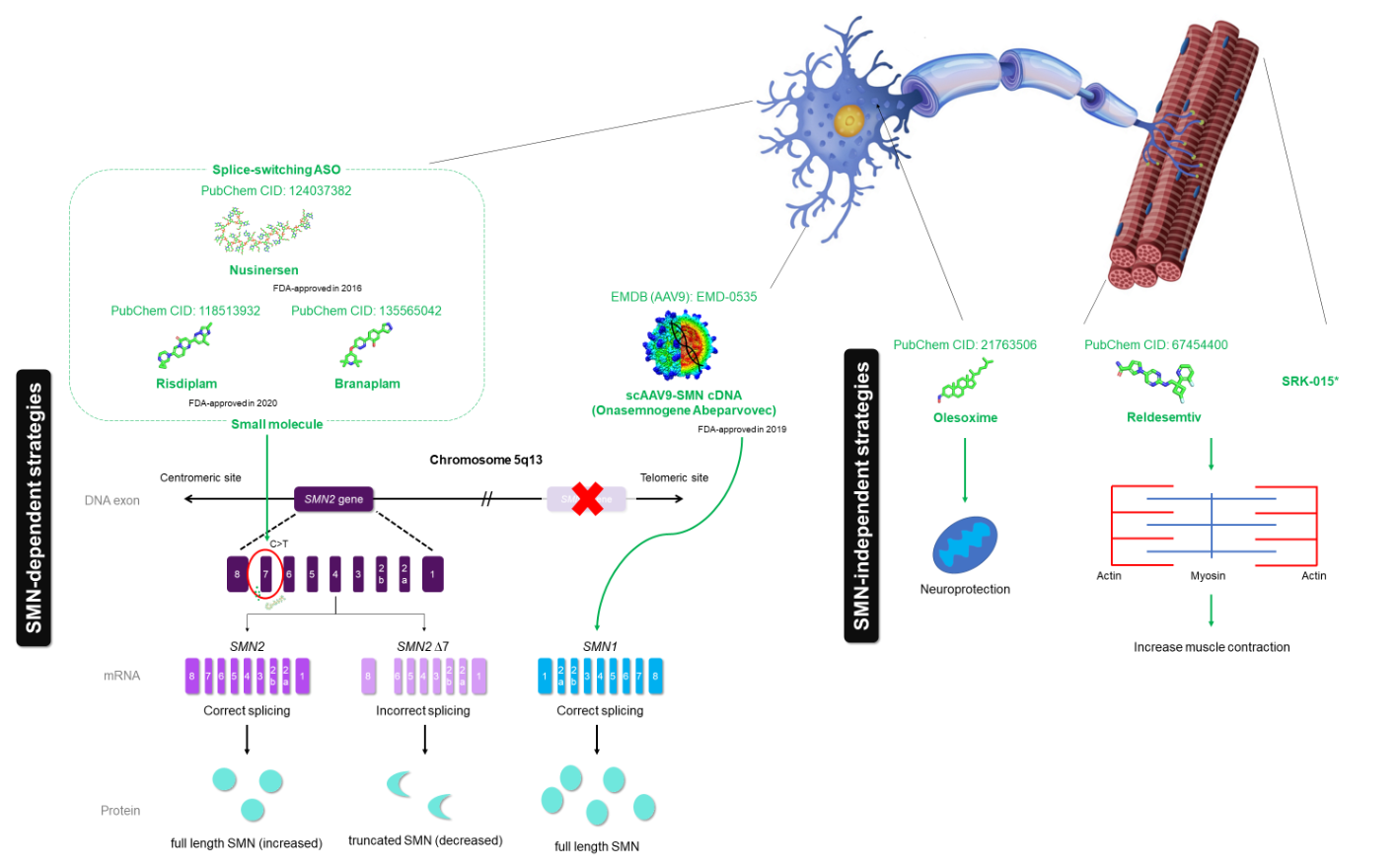 Figure 3. Therapeutic mechanism of SMA drugs, including three FDA-approved drugs (nusinersen, onasemnogene abeparvovec and the recent FDA-approved drug risdiplam) and drugs that are in clinical trials (branaplam, olesoxime, reldesemtiv and SRK-015). Nusinersen (PubChem CID: 124037382), a synthetic antisense oligonucleotide (ASO), is designed to hybridize intronic splicing silencer N1 (ISS-N1), which is heterogenous nuclear ribonucleoprotein (hnRNP) A1-dependent, to facilitate accurate splicing of SMN2 transcripts. Onasemnogene abeparvovec (no available structure) is a gene therapy that targets the SMN1 gene replacement using adenovirus vector AAV9 (EMDB: EMD-0535). Risdiplam (PubChem CID: 118513932) and branaplam (PubChem CID: 135565042) are small molecules that have the same mechanism of action as nusinersen. The red ‘X’ mark represents the deleted SMN1 gene. Other than SMN-dependent drugs, olesoxime (PubChem CID: 21763506) acts as neuroprotective compound, while reldesemtiv (PubChem CID: 67454400) and SRK-015 act as a fast skeletal muscle troponin activator (FSTA) and myostatin inhibitor, respectively, to increase muscle contraction.
Figure 3. Therapeutic mechanism of SMA drugs, including three FDA-approved drugs (nusinersen, onasemnogene abeparvovec and the recent FDA-approved drug risdiplam) and drugs that are in clinical trials (branaplam, olesoxime, reldesemtiv and SRK-015). Nusinersen (PubChem CID: 124037382), a synthetic antisense oligonucleotide (ASO), is designed to hybridize intronic splicing silencer N1 (ISS-N1), which is heterogenous nuclear ribonucleoprotein (hnRNP) A1-dependent, to facilitate accurate splicing of SMN2 transcripts. Onasemnogene abeparvovec (no available structure) is a gene therapy that targets the SMN1 gene replacement using adenovirus vector AAV9 (EMDB: EMD-0535). Risdiplam (PubChem CID: 118513932) and branaplam (PubChem CID: 135565042) are small molecules that have the same mechanism of action as nusinersen. The red ‘X’ mark represents the deleted SMN1 gene. Other than SMN-dependent drugs, olesoxime (PubChem CID: 21763506) acts as neuroprotective compound, while reldesemtiv (PubChem CID: 67454400) and SRK-015 act as a fast skeletal muscle troponin activator (FSTA) and myostatin inhibitor, respectively, to increase muscle contraction.Despite the discovery of promising therapeutic strategies, the limitations, including the treatment viability (in the case of nusinersen), long-term effects, side effects and cost, among others, are highlighted. As the drugs need to pass through the blood–brain barrier (BBB), nusinersen must be administrated locally through an intrathecal injection. This route of administration is challenging and requires sophisticated personnel and technique, such as image-guided technique, particularly for patients with scoliosis and/or spinal deformity [56]. Moreover, elevated costs of nusinersen (~USD $125,000 per injection) associated with screening and subsequent treatment (~USD $750,000 in the first year and ~USD $375,000 annually for subsequent year) place this drug among the most expensive drugs [6][57]. For the latest approved gene therapy, onasemnogene abeparvovec costs ~USD $2.125 million per injection, although only a single treatment is required for each SMA type I patient [58], while the cost of risdiplam (the most recent FDA-approved drug) is yet unknown. Additionally, as all are relatively new therapies, there are no longitudinal studies for long-term effects, although there are a plethora of studies for side effects. Therefore, a more cost-effective drug with an alternative route of administration is required for this devastating SMA.
4. Computer-Aided Drug Design (CADD)—The Open Window of Therapeutic Agents
4.1. In Silico Drug Repurposing
Drug repurposing, also known as drug repositioning, is one of the emerging potential approaches to circumvent the cost and time required for the development of an efficacious treatment [60][63]. It is defined as a process of identifying new therapeutic indications for an approved drug. Recently, with the encouragement of fast track marketing authorization procedure (FDA approvals), this approach has been widely used for rare diseases [63], including SMA [64], because it offers several benefits over the classical de novo development process of drugs. The approved drug compounds, in essence, have passed safety efficacy, allowing an omission of Phase I clinical trials [64][65].
Several studies have successfully repurposed FDA-approved drugs for SMA treatment and showed plausible in vitro activities, such as enhancing the SMN2 promoter activity, modulating SMN2 splicing and stabilizing SMN2 mRNA or SMN protein [51][64][66]. Histone deacetylase inhibitors (HDAC), including sodium butyrate, phenylbutyrate and valproic acid (VPA), among others, to date, have been explored with SMN2 promoter activity [66][67][68][69][70]. In essence, SMN-independent drugs are centred on neuroprotective and muscle enhancing approaches. In referencing the localization of the SMN protein in neuronal cells, neuroprotective drugs for other CNS diseases could be a better option to reposition for preventing and/or delaying motor neuron death in SMA. Approved neuroprotective drugs, such as riluzole, hydroxyurea and rasagiline, which modulate regulatory pathways in CNS, may be an option for SMA therapy [51][64][66].
Given the potential of the drug repurposing approach, with the combination of publicly available databases and computational methods, the in silico-based approach may provide benefits, in terms of time and cost, towards the drug discovery process by narrowing down the top hits through in silico validations [71]. Public repositories for relevant experimental and biological data, including chemical structures, gene expression, drug disease association, phenotypic traits, side effects and more, are treasure troves for in silico drug repurposing. Owing to the wealth of multi-omics data, different methods have been adopted in drug repurposing, which can be divided into two major categories: (i) drug-oriented and (ii) disease/therapy-oriented [72].
Drug-oriented drug repurposing strategies require the knowledge of cheminformatics and bioinformatics as a foundation, including drug information, chemical structures of drug and target, drug-target network, signalling or metabolic pathway and genomic information. The disease-oriented approach is only applicable if the information of the disease model is available and commonly used to study the contribution of pharmacological characteristics towards drug repositioning effort on a particular disease. The blooming of drug repurposing resources and the advances in computational sciences give rise to the development of novel algorithms/tools and approaches that are capable of capitalizing on publicly available data.
4.2. Network-Driven Drug Discovery (NDD)
Network biology epitomizes the cell as a cluster of molecules interacting with one another and aims to illustrate the emergence of cellular phenotype from the network of molecular interactions [73]. The networks can be regarded as establishing the mechanistic bridge between the constituent molecules of a cell and the phenotypes that the cells demonstrate. This perspective alone considers the cellular mechanism of disease to be materialized due to networks of pathological interactions that occur only in the disease state. In this context, drug discovery can, hence, be perceived as the search for agents that significantly disrupt these pathological networks. NDD, as a whole, aims to identify signatures of molecular perturbations; that is, collections of multiple proteins, that significantly disturb the structural integrity of the cellular networks bringing forth the targeted disease mechanism [74]. The search space of therapeutics, such as small molecules, biologics or other agents, can then be screened and narrowed down based on their ability to produce the identified perturbation signature. It should be acknowledged that the compounds of this scheme are not expected to directly bind to all proteins within the identified signature, but rather to produce a downstream, functional effect on the molecules making up the signature [75]. This approach is far removed from the traditional target-driven drug discovery that focuses on specific drug targets, whose downstream effects will significantly perturb the disease phenotype without much emphasis on cellular networks for understanding the underlying disease mechanisms.
As opposed to the canonical SMN-independent treatment based on many disease-modifying pathways, potential drug targets may be found on the periphery of the pathways using the NDD approach. A network analysis based on the two main proteins (Figure 4), SMN1 and SMN2, as protein input in GeneMANIA (https://genemania.org/) [76], has generated a network of putative interacting proteins that works in unison to bring about the phenotypes as seen in SMA. Proteins such as GEMINs [77], SNRPB [78], DDX20 [79] and PFN2 [80] appear to be highly correlated to the functioning of SMN1 and SMN2. These proteins are essential to SMN in forming macromolecular complexes (e.g., SMN-GEMINs, SMN-snRNPs) to chaperon the assembly of small nuclear ribonucleoproteins (snRNPs) that are vital to pre-mRNA splicing for producing the final SMN1 and SMN2 proteins [77]. Modulating these proteins in the cellular network within the context of SMA may serve as an opportunity to develop novel therapeutics complementary to the conventional SMN-dependent treatments in addressing the challenge of creating a robust and sustainable solution to curing SMA.
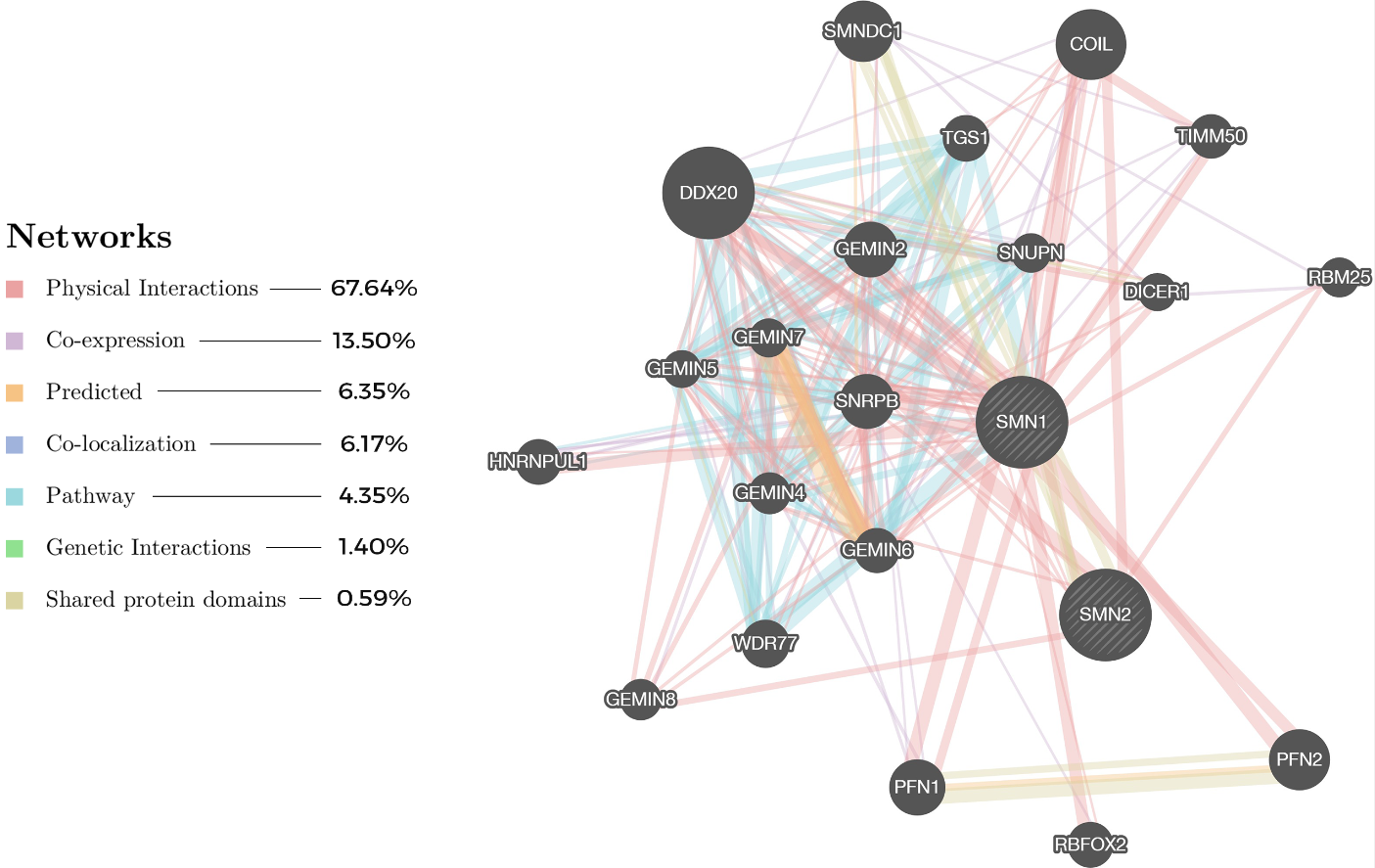 Figure 4. Protein network based on two main proteins, SMN1 and SMN2, and their respective interactions with other proteins related to SMA, generated using GeneMANIA [76] (https://genemania.org/). The most prevalent network relationship, reported by literature, among the proteins is physical interactions (pink color) at 67.64%, as visually shown by the line thickness, while the smallest belong to the shared protein domains at 0.59%.
Figure 4. Protein network based on two main proteins, SMN1 and SMN2, and their respective interactions with other proteins related to SMA, generated using GeneMANIA [76] (https://genemania.org/). The most prevalent network relationship, reported by literature, among the proteins is physical interactions (pink color) at 67.64%, as visually shown by the line thickness, while the smallest belong to the shared protein domains at 0.59%.
With the advances of network biology, the rapid growth of publicly available biomedical data and advanced computational analytics, the NDD approach, a mechanistic based approach, proposes an alternative to identify the novel target as potential SMN-independent treatment.
4.3. AI-Assisted Drug Discovery (AID)
Driven by the big data in the field of biomedical and/or healthcare, the advancement of algorithms and technology such as deep learning (DL), graphical processing units (GPUs) and Google’s tensor processing units (TPUs) enable better predictive capability by shortening the computing time [81][82]. To date, AI has been extensively adopted to support healthcare services and research. Virtual screening [83], quantitative structure-activity relationship (QSAR) [84], de novo drug design [85][86], drug repurposing [87] and chemical space visualization [88] utilized ML extensively to reduce the gap in the conventional methods in drug discovery, while DL shows promise in proposing potent drug candidates using their properties and toxicity risks [9]. Uptake from the pharmaceutical industry is still lagged, especially for rare diseases. Given the breadth of AID, we summarized the pipeline and its pre-requisites (Figure 5).
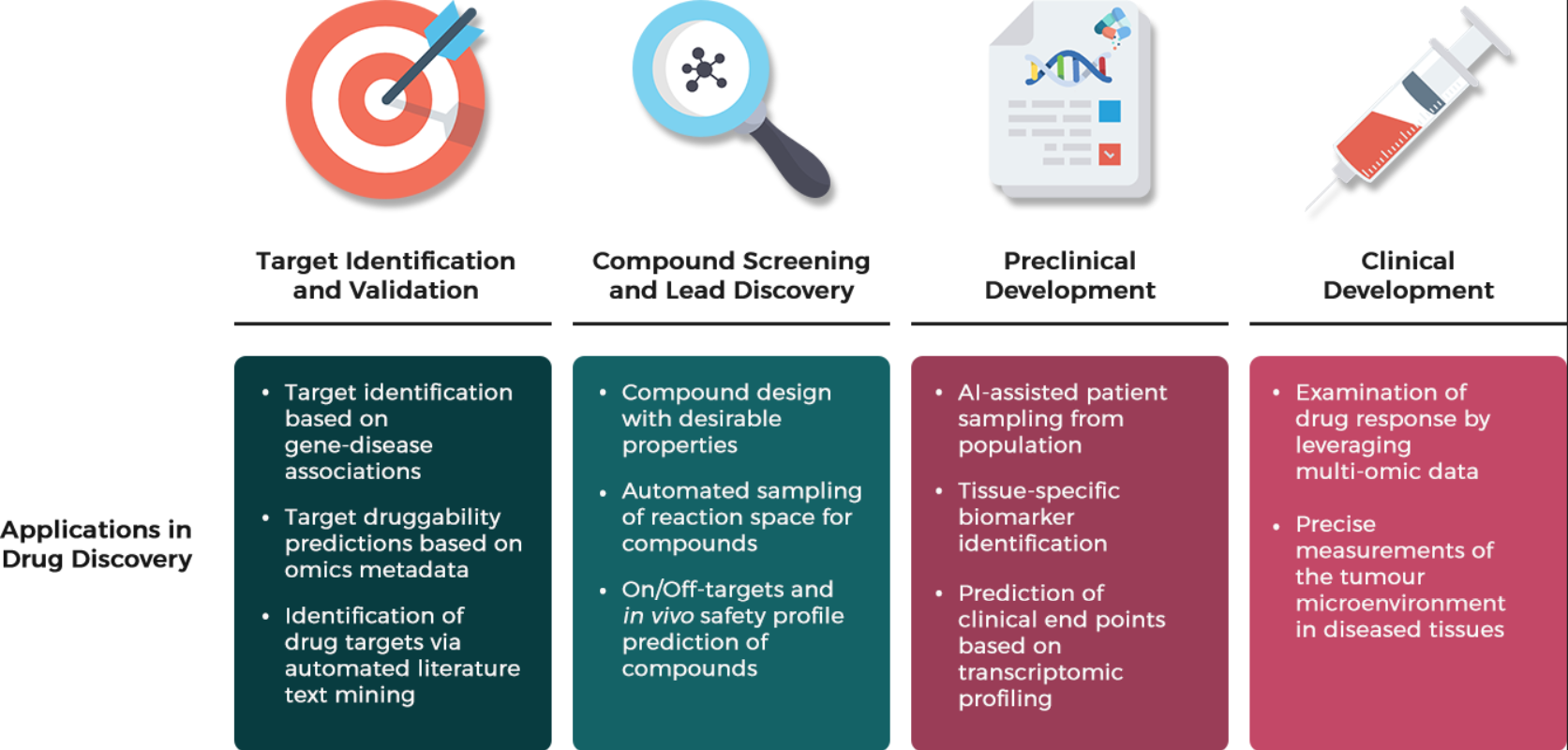 Figure 5. Machine learning applications in the drug discovery pipeline. Promising developments of pioneering ML research has brought forth unprecedented advances across various stages of the traditional drug development pipeline, especially the concept of automation in the early drug discovery process of target identification and validation & compound screening and lead discovery; relying on the domain of NLP in AI to find prospective drug targets by scanning upon thousands of relevant literature based on contextual information in research papers, and integrating AI with synthesis robots to explore unknown reaction space to search for drug candidates in which multiple chemical experiments are conducted automatically in real-time to assess the reproducibility of chemical reactions and discover new reaction outcomes. AI in the preclinical development has been a game-changer for patient selection in Phase II and III clinical trials by identifying and predicting human-relevant biomarkers of diseases, thus preventing unnecessary toxicities and side effects of consuming the experimental drugs for the designated patients [89].
Figure 5. Machine learning applications in the drug discovery pipeline. Promising developments of pioneering ML research has brought forth unprecedented advances across various stages of the traditional drug development pipeline, especially the concept of automation in the early drug discovery process of target identification and validation & compound screening and lead discovery; relying on the domain of NLP in AI to find prospective drug targets by scanning upon thousands of relevant literature based on contextual information in research papers, and integrating AI with synthesis robots to explore unknown reaction space to search for drug candidates in which multiple chemical experiments are conducted automatically in real-time to assess the reproducibility of chemical reactions and discover new reaction outcomes. AI in the preclinical development has been a game-changer for patient selection in Phase II and III clinical trials by identifying and predicting human-relevant biomarkers of diseases, thus preventing unnecessary toxicities and side effects of consuming the experimental drugs for the designated patients [89].
Through a closer inspection of AI techniques in accelerating drug discovery, there are several common machine learning methods being employed to address the challenges in two major areas of drug development: (i) design and discovery and preclinical research; and (ii) clinical research and safety monitoring.
5. Conclusions
The task of finding a successful, novel drug as treatment for common diseases is predominantly a daunting yet arduous process, which is even more challenging for a rare genetic neurological disorder such as SMA. Many research and development pharmaceutical companies and research institutions are hesitant to pursue the drug development for rare diseases due to the small market size, high cost, possibly low return and lack of information about the disease, drugs and corresponding drug targets. Recently, CADD approaches have shown promising potentials in facilitating the drug discovery process and may be able to overcome the limiting bottlenecks of its traditional counterparts. Along with the advances of the knowledge of computational biology and informatics database, the opportunities provided by drug repurposing cannot be underestimated. The interactions of a drug and a target is a critical point of drug discovery. This information aids to establish correlations between diseases and targets in order to determine the therapeutic effect of drugs on various diseases. Hence, the well-known drug–disease relationships that has been established using network biology will help accelerate the target identification and lead optimization process for pre-clinical drug development. Integrated with the domain-specific AI in the ‘chemical big data’, the novel approach could potentially serve as a panacea by increasing the efficiency of certain aspects of the drug discovery process. Despite the promising potential offered by CADD, there are several challenges, including the access of databases consisting all the approved drugs and their detailed profiles, in-depth knowledge of disease, particularly for multifaceted disease, among others to capitalize the benefit of CADD in advancing the domain of drug research and development.
References
- Cherry, J.J.; Kobayashi, D.T.; Lynes, M.M.; Naryshkin, N.N.; Tiziano, F.D.; Zaworski, P.G.; Rubin, L.L.; Jarecki, J. Assays for the Identification and Prioritization of Drug Candidates for Spinal Muscular Atrophy. Assay Drug Dev. Technol. 2014, 12, 315–341, 10.1089/adt.2014.587.
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy. Neurol. Clin. 2015, 33, 831–846, 10.1016/j.ncl.2015.07.004.
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and Characterization of a Spinal Muscular Atrophy-Determining Gene. Cell 1995, 80, 155–165, 10.1016/0092-8674(95)90460-3.
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy: A Timely Review. Arch. Neurol. 2011, 10.1001/archneurol.2011.74.
- Lefebvre, S.; Burlet, P.; Liu, Q.; Bertrandy, S.; Clermont, O.; Munnich, A.; Dreyfuss, G.; Melki, J. Correlation between Severity and SMN Protein Level in Spinal Muscular Atrophy. Nat. Genet. 1997, 16, 265–269, 10.1038/ng0797-265.
- Wurster, C.D.; Ludolph, A.C. Nusinersen for Spinal Muscular Atrophy. Ther. Adv. Neurol. Disord. 2018, 11, 175628561875445.
- Mahajan, R. Onasemnogene Abeparvovec for Spinal Muscular Atrophy: The Costlier Drug Ever. Int. J. Appl. Basic Med. Res. 2019, 9, 127, 10.4103/ijabmr.IJABMR_190_19.
- Tabet, R.; El Bitar, S.; Zaidan, J.; Dabaghian, G. Spinal Muscular Atrophy: The Treatment Approved. Cureus 2017, 10.7759/cureus.1644.
- Mak, K.-K.; Pichika, M.R. Artificial Intelligence in Drug Development: Present Status and Future Prospects. Drug Discov. Today 2019, 24, 773–780, 10.1016/j.drudis.2018.11.014.
- Otsuki, N.; Arakawa, R.; Kaneko, K.; Aoki, R.; Arakawa, M.; Saito, K. A New Biomarker Candidate for Spinal Muscular Atrophy: Identification of a Peripheral Blood Cell Population Capable of Monitoring the Level of Survival Motor Neuron Protein. PLoS ONE 2018, 13, e0201764, 10.1371/journal.pone.0201764.
- Crawford, T.O.; Pardo, C.A. The Neurobiology of Childhood Spinal Muscular Atrophy. Neurobiol. Dis. 1996, 3, 97–110, 10.1006/nbdi.1996.0010.
- Markowitz, J.A.; Tinkle, M.B.; Fischbeck, K.H. Spinal Muscular Atrophy in the Neonate. J. Obstet. Gynecol. Neonatal Nurs. 2004, 33, 12–20, 10.1177/0884217503261125.
- DiDonato, C.J.; Parks, R.J.; Kothary, R. Development of a Gene Therapy Strategy for the Restoration of Survival Motor Neuron Protein Expression: Implications for Spinal Muscular Atrophy Therapy. Hum. Gene Ther. 2003, 14, 179–188, 10.1089/104303403321070874.
- Tisdale, S.; Pellizzoni, L. Disease Mechanisms and Therapeutic Approaches in Spinal Muscular Atrophy. J. Neurosci. 2015, 35, 8691–8700, 10.1523/JNEUROSCI.0417-15.2015.
- Sumner, C.J.; Crawford, T.O. Two Breakthrough Gene-Targeted Treatments for Spinal Muscular Atrophy: Challenges Remain. J. Clin. Investig. 2018, 128, 3219–3227, 10.1172/JCI121658.
- Lunn, M.R.; Wang, C.H. Spinal Muscular Atrophy. Lancet 2008, 371, 2120–2133, 10.1016/S0140-6736(08)60921-6.
- Roberts, D.F.; Chavez, J.; Court, S.D.M. The Genetic Component in Child Mortality. Arch. Dis. Child. 1970, 45, 33–38, 10.1136/adc.45.239.33.
- Wirth, B. An Update of the Mutation Spectrum of the Survival Motor Neuron Gene (SMN1) in Autosomal Recessive Spinal Muscular Atrophy (SMA). Hum. Mutat. 2000, 15, 228–237, 10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9.
- D’Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal Muscular Atrophy. Orphanet J. Rare Dis. 2011, 6, 10.1186/1750-1172-6-71.
- Ogino, S.; Leonard, D.G.B.; Rennert, H.; Ewens, W.J.; Wilson, R.B. Genetic Risk Assessment in Carrier Testing for Spinal Muscular Atrophy. Am. J. Med Genet. 2002, 110, 301–307, 10.1002/ajmg.10425.
- Prior, T.W.; Snyder, P.J.; Rink, B.D.; Pearl, D.K.; Pyatt, R.E.; Mihal, D.C.; Conlan, T.; Schmalz, B.; Montgomery, L.; Ziegler, K.; et al. Newborn and Carrier Screening for Spinal Muscular Atrophy. Am. J. Med Genet. Part A 2010, 152A, 1608–1616, 10.1002/ajmg.a.33474.
- Schorling, D.C.; Pechmann, A.; Kirschner, J. Advances in Treatment of Spinal Muscular Atrophy—New Phenotypes, New Challenges, New Implications for Care. J. Neuromuscul. Dis. 2020, 7, 1–13, 10.3233/JND-190424.
- Arnold, W.D.; Kassar, D.; Kissel, J.T. Spinal Muscular Atrophy: Diagnosis and Management in a New Therapeutic Era. Muscle Nerve 2015, 51, 157–167, 10.1002/mus.24497.
- Campbell, L.; Potter, A.; Ignatius, J.; Dubowitz, V.; Davies, K. Genomic Variation and Gene Conversion in Spinal Muscular Atrophy: Implications for Disease Process and Clinical Phenotype. Am. J. Hum. Genet. 1997, 61, 40–50, 10.1086/513886.
- Bowerman, M.; Becker, C.G.; Yáñez-Muñoz, R.J.; Ning, K.; Wood, M.J.A.; Gillingwater, T.H.; Talbot, K. Therapeutic Strategies for Spinal Muscular Atrophy: SMN and Beyond. Dis. Models Mech. 2017, 10, 943–954, 10.1242/dmm.030148.
- Paushkin, S.; Charroux, B.; Abel, L.; Perkinson, R.A.; Pellizzoni, L.; Dreyfuss, G. The Survival Motor Neuron Protein of Schizosacharomyces Pombe. J. Biol. Chem. 2000, 275, 23841–23846, 10.1074/jbc.M001441200.
- Tariq, F.; Holcik, M.; MacKenzie, A. Spinal Muscular Atrophy: Classification, Diagnosis, Background, Molecular Mechanism and Development of Therapeutics. In Neurodegenerative Diseases; InTech: London, UK, 2013, 10.5772/53800.
- Cartegni, L.; Krainer, A.R. Disruption of an SF2/ASF-Dependent Exonic Splicing Enhancer in SMN2 Causes Spinal Muscular Atrophy in the Absence of SMN1. Nat. Genet. 2002, 30, 377–384, 10.1038/ng854.
- Kashima, T.; Manley, J.L. A Negative Element in SMN2 Exon 7 Inhibits Splicing in Spinal Muscular Atrophy. Nat. Genet. 2003, 34, 460–463, 10.1038/ng1207.
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A Single Nucleotide Difference That Alters Splicing Patterns Distinguishes the SMA Gene SMN1 from the Copy Gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183, 10.1093/hmg/8.7.1177.
- Gennarelli, M.; Lucarelli, M.; Capon, F.; Pizzuti, A.; Merlini, L.; Angelini, C.; Novelli, G.; Dallapiccola, B. Survival Motor-Neuron Gene Transcript Analysis in Muscles from Spinal Muscular-Atrophy Patients. Biochem. Biophys. Res. Commun. 1995, 213, 342–348, 10.1006/bbrc.1995.2135.
- Rad, I.A. Mutation Spectrum of Survival Motor Neuron Gene in Spinal Muscular Atrophy. J. Down Syndr. Chromosome Abnorm. 2017, 3, 1–2, 10.4172/2472-1115.1000118.
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA Type and SMN2 Copy Number Revisited: An Analysis of 625 Unrelated Spanish Patients and a Compilation of 2834 Reported Cases. Neuromuscul. Disord. 2018, 28, 208–215, 10.1016/j.nmd.2018.01.003.
- Al Dakhoul, S. Very Severe Spinal Muscular Atrophy (Type 0). Avicenna J. Med. 2017, 7, 32, 10.4103/2231-0770.197512.
- Feldkötter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative Analyses of SMN1 and SMN2 Based on Real-Time LightCycler PCR: Fast and Highly Reliable Carrier Testing and Prediction of Severity of Spinal Muscular Atrophy. Am. J. Hum. Genet. 2002, 70, 358–368, 10.1086/338627.
- Wirth, B.; Brichta, L.; Schrank, B.; Lochmüller, H.; Blick, S.; Baasner, A.; Heller, R. Mildly Affected Patients with Spinal Muscular Atrophy Are Partially Protected by an Increased SMN2 Copy Number. Hum. Genet. 2006, 119, 422–428, 10.1007/s00439-006-0156-7.
- Cuscó, I.; Barceló, M.J.; Rojas–García, R.; Illa, I.; Gámez, J.; Cervera, C.; Pou, A.; Izquierdo, G.; Baiget, M.; Tizzano, E.F. SMN2 Copy Number Predicts Acute or Chronic Spinal Muscular Atrophy but Does Not Account for Intrafamilial Variability in Siblings. J. Neurol. 2006, 253, 21–25, 10.1007/s00415-005-0912-y.
- Burghes, A.H.M.; Beattie, C.E. Spinal Muscular Atrophy: Why Do Low Levels of Survival Motor Neuron Protein Make Motor Neurons Sick? Nat. Rev. Neurosci. 2009, 10, 597–609, 10.1038/nrn2670.
- Monani, U.R.; Sendtner, M.; Coovert, D.D.; Parsons, D.W.; Andreassi, C.; Le, T.T.; Jablonka, S.; Schrank, B.; Rossol, W.; Prior, T.W.; et al. The Human Centromeric Survival Motor Neuron Gene (SMN2) Rescues Embryonic Lethality in Smn-/-Mice and Results in a Mouse with Spinal Muscular Atrophy. Hum. Mol. Genet. 2000, 9, 333–339, 10.1093/hmg/9.3.333.
- Russman, B.S. Spinal Muscular Atrophy: Clinical Classification and Disease Heterogeneity. J. Child Neurol. 2007, 22, 946–951, 10.1177/0883073807305673.
- Melki, J.; Lefebvre, S.; Burglen, L.; Burlet, P.; Clermont, O.; Millasseau, P.; Reboullet, S.; Benichou, B.; Zeviani, M.; Le Paslier, D.; et al. De Novo and Inherited Deletions of the 5q13 Region in Spinal Muscular Atrophies. Science 1994, 264, 1474–1477, 10.1126/science.7910982.
- Oskoui, M.; Darras, B.T.; De Vivo, D.C. Spinal Muscular Atrophy. In Spinal Muscular Atrophy; Elsevier: Amsterdam, The Netherlands, 2017; pp. 3–19, 10.1016/B978-0-12-803685-3.00001-X.
- Battaglia, G.; Princivalle, A.; Forti, F.; Lizier, C.; Zeviani, M. Expression of the SMN Gene, the Spinal Muscular Atrophy Determining Gene, in the Mammalian Central Nervous System. Hum. Mol. Genet. 1997, 6, 1961–1971, 10.1093/hmg/6.11.1961.
- Renvoise, B. Distinct Domains of the Spinal Muscular Atrophy Protein SMN Are Required for Targeting to Cajal Bodies in Mammalian Cells. J. Cell Sci. 2006, 119, 680–692, 10.1242/jcs.02782.
- Chaytow, H.; Huang, Y.-T.; Gillingwater, T.H.; Faller, K.M.E. The Role of Survival Motor Neuron Protein (SMN) in Protein Homeostasis. Cell. Mol. Life Sci. 2018, 75, 3877–3894, 10.1007/s00018-018-2849-1.
- So, B.R.; Zhang, Z.; Dreyfuss, G. The Function of Survival Motor Neuron Complex and Its Role in Spinal Muscular Atrophy Pathogenesis. In Spinal Muscular Atrophy; Elsevier: Amsterdam, The Netherlands, 2017; pp. 99–111, 10.1016/B978-0-12-803685-3.00006-9.
- Singh, R.N.; Howell, M.D.; Ottesen, E.W.; Singh, N.N. Diverse Role of Survival Motor Neuron Protein. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2017, 1860, 299–315, 10.1016/j.bbagrm.2016.12.008.
- Ponting, C.P. Tudor Domains in Proteins That Interact with RNA. Trends Biochem. Sci. 1997, 22, 51–52, 10.1016/S0968-0004(96)30049-2.
- Cho, S.; Dreyfuss, G. A Degron Created by SMN2 Exon 7 Skipping Is a Principal Contributor to Spinal Muscular Atrophy Severity. Genes Dev. 2010, 24, 438–442, 10.1101/gad.1884910.
- Lorson, M.A.; Lorson, C.L. SMN-Inducing Compounds for the Treatment of Spinal Muscular Atrophy. Future Med. Chem. 2012, 4, 2067–2084, 10.4155/fmc.12.131.
- Shorrock, H.K.; Gillingwater, T.H.; Groen, E.J.N. Overview of Current Drugs and Molecules in Development for Spinal Muscular Atrophy Therapy. Drugs 2018, 78, 293–305, 10.1007/s40265-018-0868-8.
- Fan, L. Survival Motor Neuron (SMN) Protein: Role in Neurite Outgrowth and Neuromuscular Maturation during Neuronal Differentiation and Development. Hum. Mol. Genet. 2002, 11, 1605–1614, 10.1093/hmg/11.14.1605.
- Martínez-Hernández, R.; Soler-Botija, C.; Also, E.; Alias, L.; Caselles, L.; Gich, I.; Bernal, S.; Tizzano, E.F. The Developmental Pattern of Myotubes in Spinal Muscular Atrophy Indicates Prenatal Delay of Muscle Maturation. J. Neuropathol. Exp. Neurol. 2009, 68, 474–481, 10.1097/NEN.0b013e3181a10ea1.
- Boyer, J.G.; Bowerman, M.; Kothary, R. The Many Faces of SMN: Deciphering the Function Critical to Spinal Muscular Atrophy Pathogenesis. Future Neurol. 2010, 5, 873–890, 10.2217/fnl.10.57.
- Pruss, R.M.; Giraudon-Paoli, M.; Morozova, S.; Berna, P.; Abitbol, J.-L.; Bordet, T. Drug Discovery and Development for Spinal Muscular Atrophy: Lessons from Screening Approaches and Future Challenges for Clinical Development. Future Med. Chem. 2010, 2, 1429–1440, 10.4155/fmc.10.228.
- Joo Young Song; Hyun Su Kim; Se-Jun Park; Jiwon Lee; Jeehun Lee; Nusinersen Administration in Spinal Muscular Atrophy Patients with Severe Scoliosis: Interlaminar Approaches at the Lumbar Level. Annals of Child Neurology 2020, 28, 49-56, 10.26815/acn.2020.00045.
- Santiago Zuluaga-Sanchez; Megan Teynor; Christopher Knight; Robin Thompson; Thomas Lundqvist; Mats Ekelund; Annabelle Forsmark; Adrian D. Vickers; Andrew Lloyd; Cost Effectiveness of Nusinersen in the Treatment of Patients with Infantile-Onset and Later-Onset Spinal Muscular Atrophy in Sweden. PharmacoEconomics 2019, 37, 845-865, 10.1007/s40273-019-00769-6.
- Zolgensma. SMA News Today. Retrieved 2021-9-3
- Duelen, R.; Corvelyn, M.; Tortorella, I.; Leonardi, L.; Chai, Y.C.; Sampaolesi, M. Medicinal Biotechnology for Disease Modeling, Clinical Therapy, and Drug Discovery and Development. In Introduction to Biotech Entrepreneurship: From Idea to Business; Springer International Publishing: Cham, Switzerland, 2019; pp. 89–128, 10.1007/978-3-030-22141-6_5.
- Prieto-Martínez, F.D.; López-López, E.; Eurídice Juárez-Mercado, K.; Medina-Franco, J.L. Computational Drug Design Methods—Current and Future Perspectives. In Silico Drug Design; Elsevier: Amsterdam, The Netherlands, 2019; pp. 19–44, 10.1016/B978-0-12-816125-8.00002-X.
- Danon, J.J.; Reekie, T.A.; Kassiou, M. Challenges and Opportunities in Central Nervous System Drug Discovery. Trends Chem. 2019, 1, 612–624, 10.1016/j.trechm.2019.04.009.
- Bagchi, S.; Chhibber, T.; Lahooti, B.; Verma, A.; Borse, V.; Jayant, R.D. In-Vitro Blood-Brain Barrier Models for Drug Screening and Permeation Studies: An Overview. Drug Des. Dev. Ther. 2019, 13, 3591–3605, 10.2147/DDDT.S218708.
- Wei Sun; Wei Zheng; Anton Simeonov; Drug discovery and development for rare genetic disorders. American Journal of Medical Genetics Part A 2017, 173, 2307-2322, 10.1002/ajmg.a.38326.
- Joseph M Hoolachan; Emma R Sutton; Melissa Bowerman; Teaching an old drug new tricks: repositioning strategies for spinal muscular atrophy. Future Neurology 2019, 14, FNL25, 10.2217/fnl-2019-0006.
- Ted T. Ashburn; Karl B. Thor; Drug repositioning: identifying and developing new uses for existing drugs. Nature Reviews Drug Discovery 2004, 3, 673-683, 10.1038/nrd1468.
- Alyssa N. Calder; Elliot J. Androphy; Kevin J. Hodgetts; Small Molecules in Development for the Treatment of Spinal Muscular Atrophy. Journal of Medicinal Chemistry 2016, 59, 10067-10083, 10.1021/acs.jmedchem.6b00670.
- Jan-Gowth Chang; Hsiu-Mei Hsieh-Li; Yuh-Jyh Jong; Nancy M. Wang; Chang-Hai Tsai; Hung Li; Treatment of spinal muscular atrophy by sodium butyrate. Proceedings of the National Academy of Sciences 2001, 98, 9808-9813, 10.1073/pnas.171105098.
- Catia Andreassi; Carla Angelozzi; Francesco Danilo Tiziano; Tiziana Vitali; Eleonora De Vincenzi; Alma Boninsegna; Marcello Villanova; Enrico Bertini; Antonella Pini; Giovanni Neri; et al. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. European Journal of Human Genetics 2003, 12, 59-65, 10.1038/sj.ejhg.5201102.
- Christina Brahe; Tiziana Vitali; Francesco D Tiziano; Carla Angelozzi; Anna Maria Pinto; Federica Borgo; Umberto Moscato; Enrico Bertini; Eugenio Mercuri; Giovanni Neri; et al. Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients. European Journal of Human Genetics 2004, 13, 256-259, 10.1038/sj.ejhg.5201320.
- L. Brichta; Jau-Tsuen Kao; Hui-Chin Wen; Kuo-Liong Chien; Hey-Chi Hsu; Shu-Wha Lin; Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Human Molecular Genetics 2003, 12, 2481-2489, 10.1093/hmg/ddg256.
- Farzin Sohraby; Milad Bagheri; Hassan Aryapour; Performing an In Silico Repurposing of Existing Drugs by Combining Virtual Screening and Molecular Dynamics Simulation. Methods in Molecular Biology 2018, 1903, 23-43, 10.1007/978-1-4939-8955-3_2.
- Guangxu Jin; Stephen T.C. Wong; Toward better drug repositioning: prioritizing and integrating existing methods into efficient pipelines. Drug Discovery Today 2013, 19, 637-644, 10.1016/j.drudis.2013.11.005.
- H. Engin; Attila Gursoy; Ruth Nussinov; Ozlem Keskin; Network-Based Strategies Can Help Mono- and Poly-pharmacology Drug Discovery: A Systems Biology View. Current Pharmaceutical Design 2014, 20, 1201-1207, 10.2174/13816128113199990066.
- Chris Fotis; Asier Antoranz; Dimitris Hatziavramidis; Theodore Sakellaropoulos; Leonidas G. Alexopoulos; Network-based technologies for early drug discovery. Drug Discovery Today 2018, 23, 626-635, 10.1016/j.drudis.2017.12.001.
- Ben Sidders; Anna Karlsson; Linda Kitching; Rubben Torella; Paul Karila; Anne Phelan; Network-Based Drug Discovery: Coupling Network Pharmacology with Phenotypic Screening for Neuronal Excitability. Journal of Molecular Biology 2018, 430, 3005-3015, 10.1016/j.jmb.2018.07.016.
- David Warde-Farley; Sylva L. Donaldson; Ovi Comes; Khalid Zuberi; Rashad Badrawi; Pauline Chao; Max Franz; Chris Grouios; Farzana Kazi; Christian Tannus Lopes; et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Research 2010, 38, W214-W220, 10.1093/nar/gkq537.
- Rebecca Eborg; Ruben J. Cauchi; GEMINs: potential therapeutic targets for spinal muscular atrophy?. Frontiers in Neuroscience 2014, 8, 325-325, 10.3389/fnins.2014.00325.
- Luke W. Thompson; Kim D. Morrison; Sally Shirran; Ewout J. N. Groen; Thomas H. Gillingwater; Catherine H. Botting; Judith E. Sleeman; Neurochondrin interacts with the SMN protein suggesting a novel mechanism for Spinal Muscular Atrophy pathology. Journal of Cell Science 2018, 131, jcs211482, 10.1242/jcs.211482.
- Frank Curmi; Ruben J. Cauchi; The multiple lives of DEAD-box RNA helicase DP103/DDX20/Gemin3. Biochemical Society Transactions 2018, 46, 329-341, 10.1042/bst20180016.
- Renske I. Wadman; Marc D. Jansen; Chantall A.D. Curial; Ewout J.N. Groen; Marloes Stam; Camiel A. Wijngaarde; Jelena Medic; Peter Sodaar; Kristel R. van Eijk; Manon M.H. Huibers; et al. Analysis of FUS, PFN2, TDP-43, and PLS3 as potential disease severity modifiers in spinal muscular atrophy. Neurology Genetics 2019, 6, e386, 10.1212/nxg.0000000000000386.
- Yann LeCun; Yoshua Bengio; Geoffrey Hinton; Deep learning. Nature 2015, 521, 436-444, 10.1038/nature14539 10.1038/nature14539.
- Erik Gawehn; Jan Alexander Hiss; J. B. Brown; Gisbert Schneider; Advancing drug discovery via GPU-based deep learning. Expert Opinion on Drug Discovery 2018, 13, 579-582, 10.1080/17460441.2018.1465407.
- Guo-Bo Li; Ling-Ling Yang; Wen-Jing Wang; Lin-Li Li; Sheng-Yong Yang; ID-Score: A New Empirical Scoring Function Based on a Comprehensive Set of Descriptors Related to Protein–Ligand Interactions. Journal of Chemical Information and Modeling 2013, 53, 592-600, 10.1021/ci300493w.
- Stefano E. Rensi; Russ B. Altman; Shallow Representation Learning via Kernel PCA Improves QSAR Modelability. Journal of Chemical Information and Modeling 2017, 57, 1859-1867, 10.1021/acs.jcim.6b00694.
- Markus Hartenfeller; Heiko Zettl; Miriam Walter; Matthias Rupp; Felix Reisen; Ewgenij Proschak; Sascha Weggen; Holger Stark; Gisbert Schneider; DOGS: Reaction-Driven de novo Design of Bioactive Compounds. PLoS Computational Biology 2012, 8, e1002380, 10.1371/journal.pcbi.1002380.
- Daniel Merk; Lukas Friedrich; Francesca Grisoni; Gisbert Schneider; De Novo Design of Bioactive Small Molecules by Artificial Intelligence. Molecular Informatics 2018, 37, 1700153, 10.1002/minf.201700153.
- Francesco Napolitano; Yan Zhao; Vânia M Moreira; Roberto Tagliaferri; Juha Kere; Mauro D’Amato; Dario Greco; Drug repositioning: a machine-learning approach through data integration. Journal of Cheminformatics 2013, 5, 30-30, 10.1186/1758-2946-5-30.
- Dmitry S. Karlov; Sergey Sosnin; Igor V. Tetko; Maxim V. Fedorov; Chemical space exploration guided by deep neural networks. RSC Advances 2019, 9, 5151-5157, 10.1039/c8ra10182e.
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of Machine Learning in Drug Discovery and Development. Nat. Rev. Drug Discov. 2019, 18, 463–477, 10.1038/s41573-019-0024-5.

