 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | vasileios atsaves | + 3986 word(s) | 3986 | 2021-08-25 08:38:46 | | | |
| 2 | Vivi Li | + 18 word(s) | 4004 | 2021-09-09 08:04:04 | | |
Video Upload Options
Immune check point blockade therapy has revolutionized the standard of cancer treatment and is credited with producing remarkable tumor remissions and increase in overall survival. This unprecedented clinical success however is feasible for a limited number of cancer patients due to resistance occurring before or during a course of immunotherapy, which is often associated with activation of oncogenic signaling pathways, co-inhibitory checkpoints upregulation or expansion of immunosuppressive regulatory T-cells (Tregs) in the tumor microenviroment (TME). Targeted therapy aiming to inactivate a signaling pathway such as the Mitogen Activated Protein Kinases (MAPKs) has recently received a lot of attention due to emerging data from preclinical studies indicating synergy with immune checkpoint blockade therapy. The dimeric transcription factor complex Activator Protein-1 (AP-1) is a group of proteins involved in a wide array of cell processes and a critical regulator of nuclear gene expression during T-cell activation. It is also one of the downstream targets of the MAPK signaling cascade.
1. Introduction
2. Activator Protein-1 (AP-1) Transcription Factors
2.1. Regulation of Immune Response by AP-1 in Genetically Modified Mice
2.2. AP-1 and T-Cell Activation
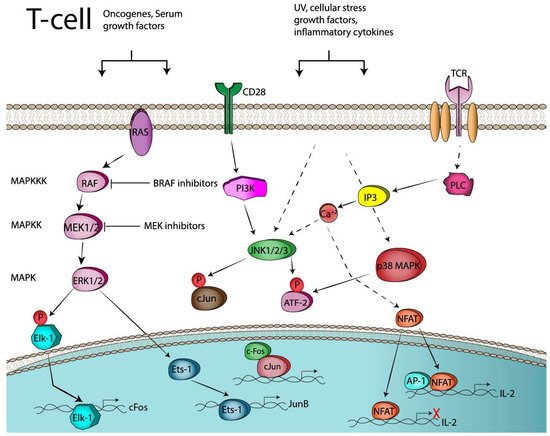
2.3. AP-1 and T-Cell Anergy or Exhaustion
3. AP-1 and Immune Checkpoint Regulation
3.1. Co-Stimulatory Molecules and AP-1 Transcription Factors
3.1.1. CD28
3.1.2. CD40/CD40L
3.2. Co-Inhibitory Molecules
3.2.1. PD-1
3.2.2. PD-L1
References
- Restifo, N.P.; Smyth, M.J.; Snyder, A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 2016, 16, 121.
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264.
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086.
- Feng, Y.; Roy, A.; Masson, E.; Chen, T.T.; Humphrey, R.; Weber, J.S. Exposure-response relationships of the efficacy and safety of ipilimumab in patients with advanced melanoma. Clin. Cancer Res. 2013, 19, 3977–3986.
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9.
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723.
- Pitt, J.M.; Vetizou, M.; Daillere, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and-extrinsic factors. Immunity 2016, 44, 1255–1269.
- Liu, C.; Peng, W.; Xu, C.; Lou, Y.; Zhang, M.; Wargo, J.A.; Chen, J.Q.; Li, H.S.; Watowich, S.S.; Yang, Y.; et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin. Cancer Res. 2013, 19, 393–403.
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016, 6, 202–216.
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88.
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235.
- Kono, M.; Dunn, I.S.; Durda, P.J.; Butera, D.; Rose, L.B.; Haggerty, T.J.; Benson, E.M.; Kurnick, J.T. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Mol. Cancer Res. 2006, 4, 779–792.
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.N.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219.
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 2013, 19, 1225–1231.
- Mimura, K.; Shiraishi, K.; Mueller, A.; Izawa, S.; Kua, L.F.; So, J.; Yong, W.P.; Fujii, H.; Seliger, B.; Kiessling, R.; et al. The MAPK pathway is a predominant regulator of HLA-A expression in esophageal and gastric cancer. J. Immunol. 2013, 191, 6261–6272.
- Brea, E.J.; Oh, C.Y.; Manchado, E.; Budhu, S.; Gejman, R.S.; Mo, G.; Mondello, P.; Han, J.E.; Jarvis, C.A.; Ulmert, D.; et al. Kinase regulation of human MHC class I molecule expression on cancer cells. Cancer Immunol. Res. 2016, 4, 936–947.
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP kinase inhibition promotes T Cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 2016, 44, 609–621.
- Lee, W.; Haslinger, A.; Karin, M.; Tjian, R. Activation of transcription by two factors that bind promoter and enhancer sequences of the human metallothionein gene and SV40. Nature 1987, 325, 368–372.
- Angel, P.; Imagawa, M.; Chiu, R.; Stein, B.; Imbra, R.J.; Rahmsdorf, H.J.; Jonat, C.; Herrlich, P.; Karin, M. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell 1987, 49, 729–739.
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136.
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859.
- Nakabeppu, Y.; Nathans, D. The basic region of Fos mediates specific DNA binding. EMBO J. 1989, 8, 3833–3841.
- Zerial, M.; Toschi, L.; Ryseck, R.P.; Schuermann, M.; Muller, R.; Bravo, R. The product of a novel growth factor activated gene, fos B, interacts with JUN proteins enhancing their DNA binding activity. EMBO J. 1989, 8, 805–813.
- Halazonetis, T.D.; Georgopoulos, K.; Greenberg, M.E.; Leder, P. C-Jun dimerizes with itself and with c-Fos, forming complexes of different DNA binding affinities. Cell 1988, 55, 917–924.
- Chiu, R.; Angel, P.; Karin, M. Jun-B differs in its biological properties from, and is a negative regulator of, c-Jun. Cell 1989, 59, 979–986.
- Schutte, J.; Viallet, J.; Nau, M.; Segal, S.; Fedorko, J.; Minna, J. Jun-B inhibits and c-fos stimulates the transforming and trans-activating activities of c-jun. Cell 1989, 59, 987–997.
- Li, B.; Tournier, C.; Davis, R.J.; Flavell, R.A. Regulation of IL-4 expression by the transcription factor Jun-B during T helper cell differentiation. EMBO J. 1999, 18, 420–432.
- Behrens, A.; Sabapathy, K.; Graef, I.; Cleary, M.; Crabtree, G.R.; Wagner, E.F. Jun N-terminal kinase 2 modulates thymocyte apoptosis and T cell activation through c-Jun and nuclear factor of activated T cell (NF-AT). Proc. Natl. Acad. Sci. USA 2001, 98, 1769–1774.
- Carrozza, M.L.; Jacobs, H.; Acton, D.; Verma, I.; Berns, A. Overexpression of the Fos B2 gene in thymocytes causes aberrant development of T cells and thymic epithelial cells. Oncogene 1997, 14, 1083–1091.
- Huse, M. The T-cell-receptor signaling network. J. Cell Sci. 2009, 122, 1269–1273.
- Linsley, P.S.; Ledbetter, J.A. The role of the CD28 receptor during T cell responses to antigen. Annu. Rev. Immunol. 1993, 11, 191–212.
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619.
- Marais, R.; Wynne, J.; Treisman, R. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell 1993, 73, 381–393.
- Hibi, M.; Lin, A.; Smeal, T.; Minden, A.; Karin, M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993, 7, 2135–2148.
- Spangler, B.; Vardimon, L.; Bosserhoff, A.K.; Kuphal, S. Post-transcriptional regulation controlled by E-cadherin is important for c-Jun activity in melanoma. Pigment. Cell Melanoma Res. 2011, 24, 148–164.
- Edmead, C.E.; Patel, Y.I.; Wilson, A.; Boulougouris, G.; Hall, N.D.; Ward, S.G.; Sansom, D.M. Induction of activator protein (AP)-1 and nuclear factor-kappaB by CD28 stimulation involves both phosphatidylinositol 3-kinase and acidic sphingomyelinase signals. J. Immunol. 1996, 157, 3290–3297.
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472.
- Macian, F.; Lopez-Rodriguez, C.; Rao, A. Partners in transcription: NFAT and AP-1. Oncogene 2001, 20, 2476–2489.
- McGuire, K.L.; Iacobelli, M. Involvement of Rel, Fos, and Jun proteins in binding activity to the IL-2 promoter CD28 response element/AP-1 sequence in human T cells. J. Immunol. 1997, 159, 1319–1327.
- Schwartz, R.H. T cell anergy. Annu. Rev. Immunol. 2003, 21, 305–334.
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499.
- Martinez, G.J.; Pereira, R.M.; Aijo, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity 2015, 42, 265–278.
- Macian, F.; Garcia-Cozar, F.; Im, S.H.; Horton, H.F.; Byrne, M.C.; Rao, A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell 2002, 109, 719–731.
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684.
- Fields, P.E.; Gajewski, T.F.; Fitch, F.W. Blocked Ras activation in anergic CD4+ T cells. Science 1996, 271, 1276–1278.
- Li, W.; Whaley, C.D.; Mondino, A.; Mueller, D.L. Blocked signal transduction to the ERK and JNK protein kinases in anergic CD4+ T cells. Science 1996, 271, 1272–1276.
- Mognol, G.P.; González-Avalos, E.; Ghosh, S.; Spreafico, R.; Gudlur, A.; Rao, A.; Damoiseaux, R.; Hogan, P.G. Targeting the NFAT: AP-1 transcriptional complex on DNA with a small-molecule inhibitor. Proc. Natl. Acad. Sci. USA 2019, 116, 9959–9968.
- Rizvi, N.A.; Mazieres, J.; Planchard, D.; Stinchcombe, T.E.; Dy, G.K.; Antonia, S.J.; Horn, L.; Lena, H.; Minenza, E.; Mennecier, B.; et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): A phase 2, single-arm trial. Lancet Oncol. 2015, 16, 257–265.
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330.
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319.
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242.
- Gross, J.A.; St John, T.; Allison, J.P. The murine homologue of the T lymphocyte antigen CD28. Molecular cloning and cell surface expression. J. Immunol. 1990, 144, 3201–3210.
- Shahinian, A.; Pfeffer, K.; Lee, K.P.; Kundig, T.M.; Kishihara, K.; Wakeham, A.; Kawai, K.; Ohashi, P.S.; Thompson, C.B.; Mak, T.W. Differential T cell costimulatory requirements in CD28-deficient mice. Science 1993, 261, 609–612.
- Foletta, V.C.; Segal, D.H.; Cohen, D.R. Transcriptional regulation in the immune system: All roads lead to AP-1. J. Leukoc. Biol. 1998, 63, 139–152.
- Granelli-Piperno, A.; Nolan, P. Nuclear transcription factors that bind to elements of the IL-2 promoter. Induction requirements in primary human T cells. J. Immunol. 1991, 147, 2734–2739.
- Rincon, M.; Flavell, R.A. AP-1 transcriptional activity requires both T-cell receptor-mediated and co-stimulatory signals in primary T lymphocytes. EMBO J. 1994, 13, 4370–4381.
- Su, B.; Jacinto, E.; Hibi, M.; Kallunki, T.; Karin, M.; Ben-Neriah, Y. JNK is involved in signal integration during costimulation of T lymphocytes. Cell 1994, 77, 727–736.
- Chatta, G.S.; Spies, A.G.; Chang, S.; Mize, G.J.; Linsley, P.S.; Ledbetter, J.A.; Morris, D.R. Differential regulation of proto-oncogenes c-jun and c-fos in T lymphocytes activated through CD28. J. Immunol. 1994, 153, 5393–5401.
- Janardhan, S.V.; Praveen, K.; Marks, R.; Gajewski, T.F. Evidence implicating the Ras pathway in multiple CD28 costimulatory functions in CD4+ T cells. PLoS ONE 2011, 6, e24931.
- Li, W.; Whaley, C.D.; Bonnevier, J.L.; Mondino, A.; Martin, M.E.; Aagaard-Tillery, K.M.; Mueller, D.L. CD28 signaling augments Elk-1-dependent transcription at the c-fos gene during antigen stimulation. J. Immunol. 2001, 167, 827–835.
- Van Kooten, C.; Banchereau, J. Functions of CD40 on B cells, dendritic cells and other cells. Curr. Opin. Immunol. 1997, 9, 330–337.
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172.
- Bishop, G.A.; Moore, C.R.; Xie, P.; Stunz, L.L.; Kraus, Z.J. TRAF proteins in CD40 signaling. Adv. Exp. Med. Biol. 2007, 597, 131–151.
- Georgopoulos, N.T.; Steele, L.P.; Thomson, M.J.; Selby, P.J.; Southgate, J.; Trejdosiewicz, L.K. A novel mechanism of CD40-induced apoptosis of carcinoma cells involving TRAF3 and JNK/AP-1 activation. Cell Death Differ. 2006, 13, 1789–1801.
- Tsytsykova, A.V.; Tsitsikov, E.N.; Geha, R.S. The CD40L promoter contains nuclear factor of activated T cells-binding motifs which require AP-1 binding for activation of transcription. J. Biol. Chem. 1996, 271, 3763–3770.
- Afford, S.C.; Ahmed-Choudhury, J.; Randhawa, S.; Russell, C.; Youster, J.; Crosby, H.A.; Eliopoulos, A.; Hubscher, S.G.; Young, L.S.; Adams, D.H. CD40 activation-induced, Fas-dependent apoptosis and NF-kappaB/AP-1 signaling in human intrahepatic biliary epithelial cells. FASEB J. 2001, 15, 2345–2354.
- Baccam, M.; Woo, S.Y.; Vinson, C.; Bishop, G.A. CD40-mediated transcriptional regulation of the IL-6 gene in B lymphocytes: Involvement of NF-kappa B, AP-1, and C/EBP. J. Immunol. 2003, 170, 3099–3108.
- Vanden Bush, T.J.; Bishop, G.A. TLR7 and CD40 cooperate in IL-6 production via enhanced JNK and AP-1 activation. Eur. J. Immunol. 2008, 38, 400–409.
- D’Aversa, T.G.; Eugenin, E.A.; Berman, J.W. CD40-CD40 ligand interactions in human microglia induce CXCL8 (interleukin-8) secretion by a mechanism dependent on activation of ERK1/2 and nuclear translocation of nuclear factor-kappaB (NFkappaB) and activator protein-1 (AP-1). J. Neurosci. Res. 2008, 86, 630–639.
- Arasanz, H.; Gato-Cañas, M.; Zuazo, M.; Ibañez-Vea, M.; Breckpot, K.; Kochan, G.; Escors, D. PD1 signal transduction pathways in T cells. Oncotarget 2017, 8, 51936–51945.
- Sheppard, K.A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004, 574, 37–41.
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017, 355, 1428–1433.
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029.
- Quigley, M.; Pereyra, F.; Nilsson, B.; Porichis, F.; Fonseca, C.; Eichbaum, Q.; Julg, B.; Jesneck, J.L.; Brosnahan, K.; Imam, S.; et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat. Med. 2010, 16, 1147–1151.
- Xiao, G.; Deng, A.; Liu, H.; Ge, G.; Liu, X. Activator protein 1 suppresses antitumor T-cell function via the induction of programmed death 1. Proc. Natl. Acad. Sci. USA 2012, 109, 15419–15424.
- Chen, L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004, 4, 336–347.
- Zerdes, I.; Matikas, A.; Bergh, J.; Rassidakis, G.Z.; Foukakis, T. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: Biology and clinical correlations. Oncogene 2018, 37, 4639–4661.
- Mathas, S.; Hinz, M.; Anagnostopoulos, I.; Krappmann, D.; Lietz, A.; Jundt, F.; Bommert, K.; Mechta-Grigoriou, F.; Stein, H.; Dorken, B.; et al. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappa B. EMBO J. 2002, 21, 4104–4113.
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618.
- Fang, W.; Zhang, J.; Hong, S.; Zhan, J.; Chen, N.; Qin, T.; Tang, Y.; Zhang, Y.; Kang, S.; Zhou, T.; et al. EBV-driven LMP1 and IFN-gamma up-regulate PD-L1 in nasopharyngeal carcinoma: Implications for oncotargeted therapy. Oncotarget 2014, 5, 12189–12202.
- Jiang, X.; Zhou, J.; Giobbie-Hurder, A.; Wargo, J.; Hodi, F.S. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin. Cancer Res. 2013, 19, 598–609.
- Sumimoto, H.; Takano, A.; Teramoto, K.; Daigo, Y. RAS-mitogen-activated protein kinase signal is required for enhanced PD-L1 expression in human lung cancers. PLoS ONE 2016, 11, e0166626.
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165.


