 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vanessa D’Antongiovanni | + 4135 word(s) | 4135 | 2020-07-20 08:18:34 | | | |
| 2 | Camila Xu | -107 word(s) | 4028 | 2020-10-27 08:59:51 | | |
Video Upload Options
Adenosine is a purine nucleoside, resulting from the degradation of adenosine triphosphate (ATP). Under adverse conditions, including hypoxia, ischemia, inflammation, or cancer, the extracellular levels of adenosine increase significantly. Once released, adenosine activates cellular signaling pathways through the engagement of the four known G-protein-coupled receptors, adenosine A1 receptor subtype (A1), A2A, A2B, and A3. These receptors, expressed virtually on all immune cells, mitigate all aspects of immune/inflammatory responses. These immunosuppressive effects contribute to blunt the exuberant inflammatory responses, shielding cells, and tissues from an excessive immune response and immune-mediated damage.
1. Introduction
The inflammatory process is a protective response aimed at stemming external insult (i.e., pathogens, toxic compounds, and irradiation) to preserve tissue integrity [1]. However, when inflammation goes beyond this protective purpose, and due to altered molecular mechanisms, there is a chronicization of the inflammatory process, thus creating the conditions for the development of microniches favorable for the development of neoplasia [1].
In this regard, a number of studies well described a causal link between the presence of a chronic inflammatory process and cancer development [2–6]. Indeed, it has been observed that about 20% of neoplastic diseases are observed in patients with a history of chronic inflammatory diseases [2–6]. In this regard, epidemiological investigations pointed out that patients with ulcerative colitis (UC) and Crohn’s disease (CD) are 3–6 times more likely to develop colorectal cancer (CRC) than the general population [7–9], thus corroborating the presence of a “thin red line” between an exasperated immune system activity and a neoplastic drift of the affected tissues.
Over the years, there has been an evolution of the concept of tumor microenvironment, initially considered as a tissue merely surrounding the tumor mass, indicating the cells composing the neoplastic niche participate actively in supporting the growth of carcinogen-altered cells to form focal lesions [10–13].
In the early stages of tumor development, the different immune cell populations, such as macrophages, neutrophils, mast cells, dendritic cells (DCs), and lymphocytes, intervene promptly with anti-tumor responses aimed at erasing the cancer cells [14]. Of note, in this context, it is conceivable that some neoplastic cell variants can acquire a less immunogenic phenotype thus escaping to immune detection [14]. Consequently, such immune-resistant selected clones start to release massively a plethora of chemotactic factors leading to the recruitment of immune cells within the neoplastic environment [15,16]. This is a crucial step in the neoplastic onset and development since a perverse partnership is created between cancer cells and the neo-infiltrating immune cells, through a paracrine and a cell-cell contact signaling, determining a phenotypical reorganization of immune cell population with a loss of their anti-tumorigenic functions [17,18]. In this context, the immune cells, releasing various cytokines and chemokines as well as oncogenic mediators (i.e., nitric oxide and growth factors), affect cell proliferation, death, and senescence, interfering also on DNA mutation rates and methylation, or the angiogenic process [19].
In this context, several authors provided interesting evidence about the involvement of adenosine, a retaliatory metabolite resulting from the degradation of adenosine triphosphate (ATP), in neoplasia onset and progression [20–24]. Under physiological conditions, low levels of adenosine are detectable in the interstitial fluids of unstressed tissues [25–27], whereas a marked increase of extracellular adenosine levels is observed under adverse conditions, including hypoxia, ischemia or inflammation [23,27,28]. Such an increase in the adenosine levels represents one of the pro-resolutive mechanisms aimed at suppressing and extinguishing an exuberant inflammatory reaction once its main task is attained [29]. It is worth to note that the prolonged persistence of high adenosine concentrations acquires detrimental features, triggering, and maintaining an immunosuppressed milieu, an ideal context for neoplasia onset and development [20,23,30].
Indeed, the marked presence of adenosine in the tumor microenvironment plays a critical role in shaping the generation of this niche, eliciting the repolarization toward an immunosuppressive phenotype for macrophages, DCs and neutrophils, with a contextual suppression of effector T cells and an expansion of regulatory T (Treg) cells [23,30]. In parallel, adenosine contributes to tumor growth, exerting a direct proliferative effect on neoplastic cells and sustaining either the neoangiogenic process and the extracellular matrix remodeling [31,32]. It is worth noting that such increased extracellular adenosine concentrations are the results of specific genetic alterations occurring during tumor progression [33]. Indeed, it has been well described that several tumors displayed an altered purine metabolism, characterized by a magnification of the molecular mechanisms facilitating the production of adenosine and by inhibition of the systems deputed to the degradation, thus creating an environment particularly rich in adenosine, suitable for cancer onset, development and spread [23,33].
2. Adenosine System: Enzymes, Transporters, and Receptors
Under physiological conditions, adenosine is present at low levels in both the intracellular and extracellular space [34]. In these conditions, the main source of intracellular adenosine is S-adenosylhomocysteine via S-adenosylhomocysteine hydrolase [35]. Extracellularly, adenosine results mainly by the degradation of ATP and adenosine diphosphate (ADP) to adenosine monophosphate (AMP), and then converted to adenosine, operated by the ectonucleotidases, CD39 and CD73, respectively [36] (Figure 1). The extracellular and intracellular levels of adenosine are finely tuned by the activity of the nucleoside transporters [29], classified into (1) equilibrative nucleoside transporters (ENT1, ENT2, ENT3, and ENT4), which transport nucleosides across cell membranes in either direction, based on concentration gradients [37–39] and (2) concentrative nucleoside transporters (CNT1, CNT2, and CNT3), which shunt extracellular adenosine into the intracellular space against their concentration gradient, exploiting the sodium ion gradient across cellular membranes as a source of energy [37–39]. Another critical checkpoint in the regulation of adenosine levels is represented by the expression and activity of adenosine deaminase (ADA), a key enzyme involved in the degradation of adenosine into inosine [35] (Figure 1).
Under pathological conditions, including hypoxia, ischemia, inflammation, or cancer, the concentration of adenosine increases rapidly as a consequence of massive extracellular dephosphorylation of ATP, mediated sequentially by CD39 and CD73 [40]. In this regard, several authors identified CD73 as a critical checkpoint in the regulation of extracellular adenosine levels and, consequently, in the control of receptor stimulation either under physiological or pathological conditions [41]. Of note, adenosine can also be generated through a non-canonical pathway by which nicotinamide adenine dinucleotide (NAD+)-glycohydrolase/CD38 enzyme axis converts extracellular NAD+ into adenosine diphosphate ribose (ADPR) [42]. ADPR is then metabolized by CD203a into AMP and then converted by CD73 into adenosine [42]. As described previously, also under adverse conditions the extracellular adenosine can be re-up taken into the cells through the nucleoside transporters, or converted into inosine by ADA [35–37].
Once released, the extracellular adenosine activates cellular signaling pathways through the engagement of the four known G-protein-coupled receptors, adenosine A1 receptor subtype (A1), A2A, A2B, and A3 [43]. A1 and A3 receptors are coupled with Gi, Gq, or Go proteins, whereas A2A and A2B are coupled to Gs or Gq proteins (Figure 1). The stimulation of A1 and A3 receptors can also trigger the release of calcium ions from intracellular stores, as well as A2B receptor stimulation can also activate phospholipase C via Gq [44,45]. Of note, all the adenosine receptors are coupled to MAPK/ERK signaling pathways [44] (Figure 1). In addition, adenosine can also exert receptor-independent effects, via less defined intracellular mechanisms, including adenosine kinase, S-adenosylhomocysteine hydrolase systems, and AMP-activated protein kinase (AMPK) [46–48].
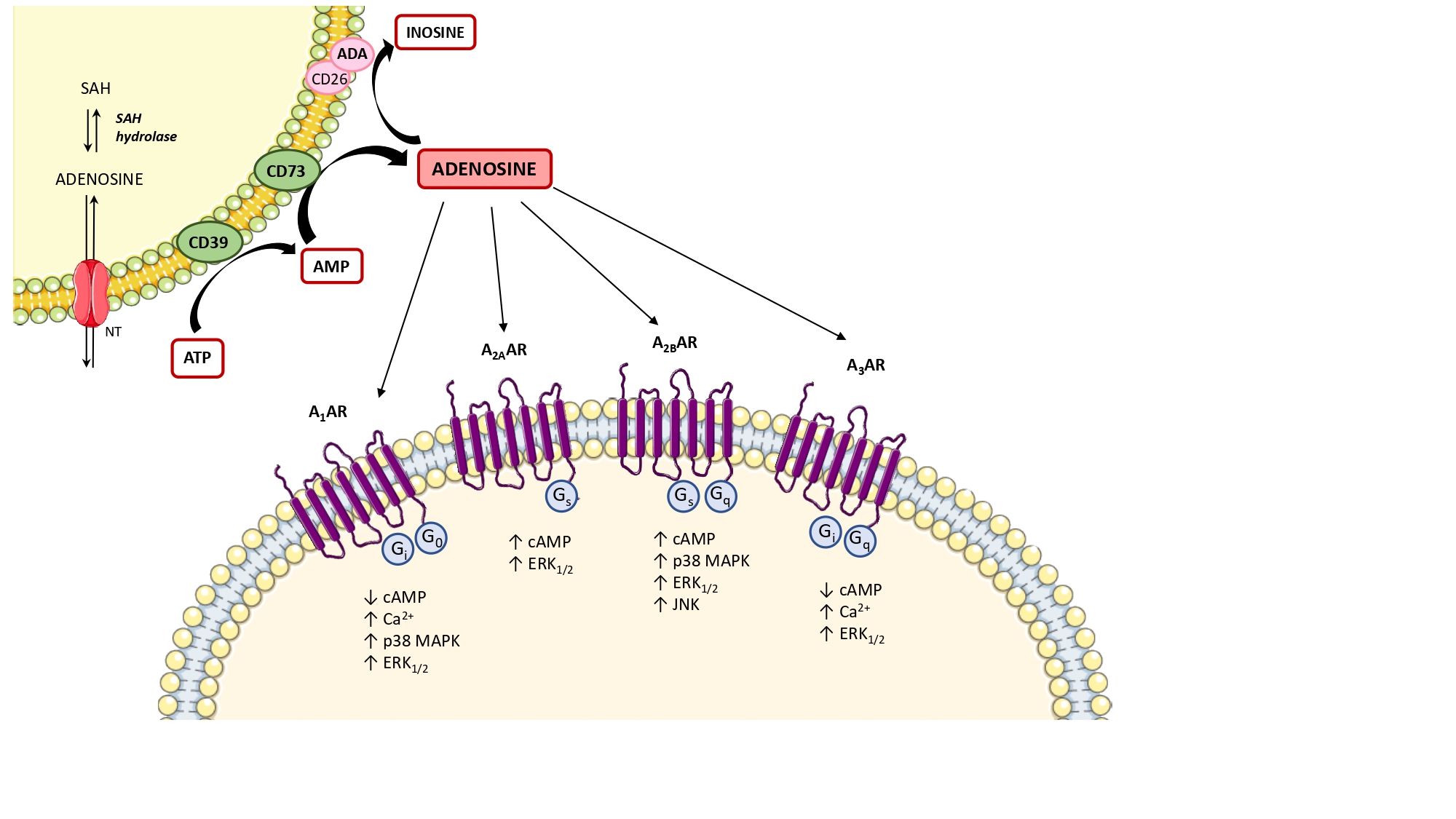
Figure 1. Diagram showing the main step of adenosine biosynthesis and catabolism and the second messenger pathways coupled to adenosine receptors. The main source of intracellular adenosine is S-adenosylhomocysteine (SAH) via S-adenosylhomocysteine hydrolase (SAH hydrolase) and then released into the extracellular space via nucleoside transporters (NTs). The extracellular adenosine is mainly produced by the ectonucleotidases, CD39 and CD73. Excess adenosine is irreversibly deaminated to inosine by the enzyme adenosine deaminase (ADA). In the extracellular space, adenosine can bind to four different G protein-coupled adenosine receptors that either inhibit (mediated by A1 and A3 adenosine receptors) or stimulate (mediated by A2A and A2B adenosine receptors) adenylyl cyclase activity and cAMP production in the cell. All adenosine receptors couple to MAPK pathways, including ERK1/2 and p38 MAPK. ↑: increase; ↓: decrease; A1AR: A1 adenosine receptor; A2AAR: A2A adenosine receptor; A2BAR: A2B adenosine receptor; A3AR: A3 adenosine receptor; ATP: adenosine triphosphate; Ca2+: calcium ions; cAMP: cyclic AMP; ERK1/2: extracellular signal-regulated kinase 1/2; JNK: JUN N‑terminal kinase; MAPK: mitogen-activated protein kinase.
3. Adenosine System and Inflammatory Bowel Diseases (IBDs)
Inflammatory bowel diseases (IBDs), comprising mainly UC and CD, are chronic intestinal inflammatory disorders, characterized by excessive inflammation of the digestive tract, associated with thrombophilia and heightened risk of developing CRC [49]. Over the years, increasing evidence pointed out that the risk of cancer development in IBD patients is time-dependent and can increase by 2% by 10 years, 8% by 20 years, and 18% by 30 years [49,50]. The risk factors for CRC among IBD patients include severe inflammation, coexisting primary sclerosing cholangitis (PSC), family history of sporadic CRC, and age of colitis onset [49–51]. In particular, the risk of cancer in CD is controversial and, compared with UC, the risk is modest [52]. Based on these premises, research efforts have been focused on a better characterization of the molecular events determining a neoplastic degeneration of inflamed tissues in IBD patients, pointing out particular attention to the role exerted by adenosine system in the modulation of long-standing inflammation before the onset of tumorigenesis.
The majority of available pre-clinical studies have been performed using the dextran sulfate sodium (DSS)-induced colitis model, a widely used murine model, closely resembling the human UC, which allows a careful evaluation about the innate immune mechanisms involved in the development of bowel inflammation [53]. This model is based on the continuous administration in drinking water with DSS, a synthetic polymer of sulfated polysaccharides, leading to a reproducible acute inflammatory process, limited to the colon. Macroscopically, DSS administration is characterized by bloody diarrhea, intestinal inflammation and ulcerations, a marked body weight loss, and shortening of colonic length [53]. Histologically, this model is characterized by hyperosmotic damage toward the intestinal epithelial layer, which elicits a marked immune response in the host. In particular, the DSS colitis displayed a cytokine profile mainly characterized by a mixed T-helper type 2 (Th2) and Th17 immune paradigm [54].
Over the years, evidence has well described the role of adenosine as a potent endogenous immunoregulatory agent, able to blunt the exuberant inflammatory responses, shielding cells and tissues from an excessive immune response and immune-mediated damage [55,56]. Following an acute injury, a series of molecular mechanisms are put in place to increase the extracellular levels of adenosine, to elicit an efficient specific immune response aimed at restoring the tissue homeostasis [56]. In the late stage of inflammation, adenosine contributes relevantly to the resolution of inflammation, both repolarizing toward an anti-inflammatory phenotype of several innate immune cell populations (i.e., macrophages, DCs, neutrophils) as well as counteracting the acquired immune cell responses, acting directly on the T effector cells and spurring the expansion and the functions of immunosuppressive Treg cells [56].
In this regard, studies performed on the pre-clinical DSS mouse model provided interesting evidence about the relevance of the adenosine system in orchestrating the immune responses (Figure 2). Siegmund et al. reported that an increase of endogenous adenosine levels, via pharmacological blockade of adenosine kinase by the selective inhibitor GP515, ameliorates DSS-induced colitis blunting the proinflammatory cytokine synthesis as well as suppressing interferon (IFN)-γ in colonic tissue [57]. Subsequently, Pallio and colleagues reported that adenosine, through A2A receptor engagement, counteracted the expression of inflammatory cytokines, such as interleukin (IL)-1β and tumor necrosis factor (TNF), ameliorating significantly also the clinical features [58]. On the same line, Selmeczy et al. (2007) provided evidence about an ameliorative effect of CGS 21680, an A2A receptor agonist, in the early stage of DSS-induced colitis [59] (Figure 2).
With regard to A2B receptors, Kolachala et al. observed an over-expression of this receptor subtype in mice treated with DSS, demonstrating that such an increase was mediated by the activation of the TNF signaling pathway [60,61] (Figure 2). A relevant role of A2B receptors in modulating the acute inflammatory phase of DSS colitis was described by Frick et al. (2009). Indeed the authors reported that the A2B receptor engagement blunted the production of pro-inflammatory cytokines, such as IFN-γ, IL-1β, and IL-12, sparing an anti-inflammatory activity mediated mainly by IL-10 [62]. The administration of PSB1115, a selective A2B receptor inhibitor, resulted in an increase in the severity of DSS colitis, thus corroborating a critical role of A2B receptors in shaping the immune cell activity in a model of DSS-induced colitis [62]. In line with this view, Aherne et al. demonstrated that the genetic ablation of A2B receptors was associated with increased severity of DSS colitis along with the loss of intestinal epithelial barrier functions [63]. Such alterations were mitigated by the treatment with selective A2B receptor agonist, BAY-606583 [63] (Figure 2).
In the last years, increasing interest has been addressed to the pharmacological modulation of A3 receptors as a viable way to manage several immuno-mediated inflammatory diseases, including IBDs [29]. In this regard, it has been observed that the genetic ablation of the A3 receptor protected against DSS colitis effects [64]. By contrast, the pharmacological stimulation of A3 receptors with IB-MECA resulted effective in protecting against colitis through the reduction of inflammatory cytokine (i.e., IL-1β, IL-6, and IL-12) and chemokine (i.e., macrophage inflammatory protein (MIP)-1α and MIP-2) levels in colonic tissues [65] (Figure 2). On the same line, other authors highlighted the beneficial effect of 2-Cl-IB-MECA administration in DSS-treated mice, with a decrease of the pro-inflammatory responses through the inhibition of NF-kB signaling pathways [66]. Recently, it has been observed that the reduction of A3 receptor expression, via the treatment with miR-206-agomir (a microRNA analog), triggers the activation of pro-inflammatory NF-κB signaling pathways in DSS animal model [67], thus corroborating a “brake role” for A3 receptors on the inflammatory process. In support of this view, Ren et al. reported that the pharmacological activation of A3 receptors with 2-Cl-IB-MECA exerted an anti-inflammatory effect in human colonic epithelial cells, blunting the NF-κB signaling pathways along with a significant reduction of IL-8 and IL-1β production [68] (Figure 2).
Nowadays, some clinical studies have provided evidence about the anti-inflammatory effects of adenosine and its receptors in human colitis (Figure 2). For instance, a recent paper, by Tian and collaborators, reported that adenosine, through the activation of A2A receptors, exerts a protective role in colonic inflammation in UC patients through the inhibition of NF-κB signaling pathway and the transcription of pro-inflammatory cytokine genes, including IFN-γ, IL-1β and IL-8 [69] (Figure 2). Other authors paid attention to the expression of A2B receptors in human UC. Kolachala et al. observed an up-regulation of this receptor subtype in colonic tissues from a large cohort of UC patients, highlighting that A2B receptor expression is modulated during inflammation [60,61] (Figure 2). With regard for the A3 receptor, Wu et al. observed that UC patients are characterized by an up-regulation of microRNA-206 (miR-206), which directly inhibits the expression of A3 receptors, with the consequent triggering of the pro-inflammatory response [67]. In line with this view, a more recent study confirmed the contribution of this receptor subtype in curbing the intestinal inflammation by inhibiting the production of TNF and IL-1β, along with an inhibition of NF-κB signaling pathway [70] (Figure 2).
Based on these premises, clinical and pre-clinical studies demonstrated that in the presence of intestinal inflammation adenosine represents a pro-resolutive mediator, acting as a modulator of the immune cellular responses to shield tissues from an excessive inflammatory response and immune-mediated damage. Indeed, adenosine, through the activation of specific receptors, inhibits the production of pro-inflammatory cytokines, including IFN-γ, IL-1β, IL-8, and IL-12, thus contributing relevantly to the resolution of inflammation. It is worth to note that the persistence of marked adenosine concentrations can become detrimental to tissues, triggering the secretion of immunosuppressive cytokines, such as IL-10 and transforming growth factor (TGF)-β from immune cells, overwhelming the immunosurveillance towards neoplastic cells.
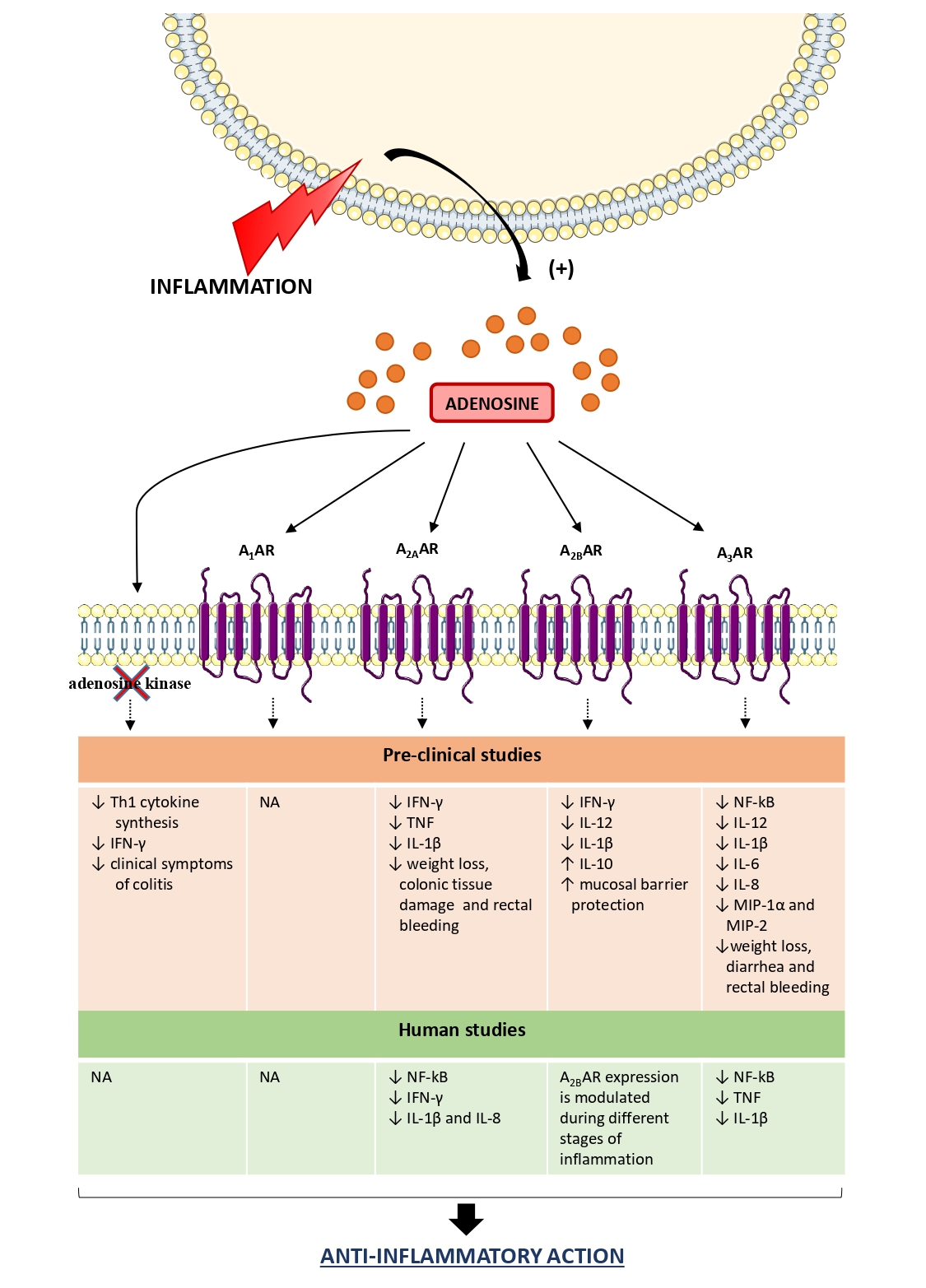
Figure 2. Role of adenosine receptors in the modulation of the inflammatory response in IBDs. In the presence of intestinal inflammation, the extracellular levels of adenosine increase significantly to restore tissue homeostasis. In this context, adenosine exerts an anti-inflammatory action, thus shielding cells and tissues from an excessive inflammatory response and immune-mediated damage. ↑: increase; ↓: decrease; A1AR: A1 adenosine receptor; A2AAR: A2A adenosine receptor; A2BAR: A2B adenosine receptor; A3AR: A3 adenosine receptor; IL: interleukin; INF- γ: interferon-γ; MIP: macrophage inflammatory protein; NA: not available; Th1: T-helper type 1; TNF: tumor necrosis factor.
4. Adenosine System and Immune Cell Interaction in Colitis-Associated Cancer Pathogenesis
Colitis-associated cancer (CAC) is a type of CRC preceded by clinically detectable IBD [71]. It is well known that long-standing enteric inflammation triggers chronic relapsing intestinal damage, thus creating a tumor-prone microenvironment [72]. In this context, adenosine exerts a critical role in tumor onset and progression, participating in the generation of an immunosuppressive and pro-angiogenic environment [23]. However, the molecular mechanisms through which the adenosine system participates in the various stages of transition from a chronic inflammatory context to the neoplastic transformation process remain poorly characterized. In this regard, the pre-clinical azoxymethane (AOM)/DSS mouse model represents a useful tool to deepen the mechanisms underlying the onset of CAC to define novel therapeutic approaches [73]. Indeed, the murine model combining the AOM, a colonic genotoxic carcinogen, with DSS, a colitis inducer, shortens the latency time for induction of CRC leading to the aberrant crypt foci-adenoma-carcinoma sequence that occurs in human CAC [73].
During cancer pathogenesis, the extracellular levels of adenosine increase significantly, reaching the micromolar range [74,75]. In this regard, an in situ microdialysis study performed in colonic specimens from human and mouse, reported a 20-fold increase of extracellular adenosine when compared to normal tissues [20]. The persistence of increased adenosine concentrations beyond its anti-inflammatory and pro-resolutive task, becomes detrimental for the tissues, generating an immunosuppressed milieu useful for the onset and development of neoplastic cells [30]. Several investigations highlighted that such extracellular adenosine accumulation results from specific genetic alterations occurring during the carcinogenic process [33].
Increasing evidence demonstrates an increase in the expression and/or activity of CD39 and CD73 in tumor endothelial cells as well as in immune cells (i.e., Treg cells, myeloid-derived suppressor cells (MDSCs), macrophages, DCs, and Th17 cells) isolated from patients with CRC [76–80]. Such increased expression and activity of the CD39-CD73 axis leads to a chronic presence of high adenosine levels in the tumor microenvironment (TME), impairing markedly the anti-tumor immune response and thus paving the way to cancer cell proliferation and angiogenic process. For this reason, the concomitant inhibition of CD39 and CD73 activity could represent an innovative pharmacological strategy in cancer therapy, aimed at counteracting tumor growth by awakening the anti-tumor immunity. Of note, the prolonged and persistent condition of inflammation and hypoxia, commonly observed in CAC patients, are considered the most frequent inducers of CD39 and CD73 expression and activity [81]. In this regard, several studies have reported that the hypoxia-inducible factor (HIF)-1α triggers an increase of CD39 and CD73 activity, thus impairing the T cell functions via the generation of high concentrations of adenosine [82–87].
In parallel, it has been observed a down-regulation of the catabolic enzyme ADA and its cofactor CD26 (also known as dipeptidyl peptidase 4) [88–90] as well as the reduction of adenosine kinase activity [91] in the cancer microenvironment.
Once released in TME, extracellular adenosine exerts a potent immunosuppressive and cancer growth-promoting effect interacting with several immune cell populations [21]. In particular, adenosine, beyond to counteract the effector T cell functions mainly via the engagement of the A2A receptors, inhibits mononuclear phagocyte cell differentiation and maturation as well as contributes to the angiogenetic processes and matrix remodeling environment, suitable for cancer growth [92–95]. Of note, the effect of adenosine on the immune system depends on its bioavailability and receptors engaged in immune cells present within TME. For instance, the activations of A2A receptors spur the Treg functions, via cAMP/protein kinase cAMP-dependent (PKA) pathways, thus promoting immune-suppressive effects [96–100] (Figure 3). In support of this view, other studies reported also that A2A receptor activation prevents natural killer (NK) cell activation, maturation, and cytotoxicity [92,99].
The marked presence of adenosine in the TME, engaging the A2A receptors expressed on macrophages and DCs, stimulates the IL-10 release and counteracts the IL-12 production, with subsequent impairment of T cell priming and the suppression of antitumor immune responses [96,100,101] (Figure 3). Of note, in several murine cancer models, including the CAC model, it has been observed a potent antitumor effect exerted by IL-12, via the reduction of distant metastases formation and prolonging significantly the survival of mice allowing an efficient anti-tumor response [102,103].
Increasing attention has been paid to the role of A2B receptor in controlling immune cell-mediated responses. In this regard, Ryzhov et al. reported that the activation of this receptor subtype promoted the expansion of MDSCs, an immature subset of myeloid cells, which express high levels of CD73, thus contributing to a further generation of adenosine with consequent increase of the immunosuppressive environment [104] (Figure 3). In parallel, it has been observed that the engagement of A2B receptors contributes to the generation of an immunosuppressive milieu, stimulating the Treg differentiation and proliferation along with an increase in IL-10 production [105,106]. In the TME, the engagement of the A2B receptors expressed on macrophages, elicits the secretion of vascular endothelial growth factor (VEGF) from these cells thus playing a relevant role in tumor angiogenesis [106] (Figure 3). In support of this view, the pharmacological blockade of A2B receptors with the selective antagonist PSB1115, reduces the release of VEGF and the number of tumor-infiltrating MDSCs in a mouse melanoma model, thus corroborating the pro-angiogenic and immunosuppressive effects of this receptor subtype [106].
This evidence was corroborated by further studies reporting that the extracellular adenosine, via A2A and A2B receptor engagement, counteracted the neutrophil activation as well as their adhesion to the endothelial cells, participating to the generation of an immunosuppressive and proangiogenic milieu [104,107,108].
Interestingly, Harish and colleagues demonstrated that the activation of A3 receptors expressed on NK cells increases their antitumor activity [109]. Recently, it has been also observed that the engagement of A1 and A3 receptors on neutrophils mediate multiple immunosuppressive effects, including the inhibition of oxidative burst response [110] (Figure 3).
At present, clinical evidence about the role of the adenosine system in the molecular mechanisms underlying the inflammation-associated CRC pathogenesis are scanty. An over-expression of CD39 and CD73 has been reported in Treg and MDSCs isolated from CRC patients [77,111], suggesting that tumor-infiltrating Treg and MDSCs are likely an important source of extracellular adenosine that contributes to the tumor immune escape. In the same study emerged that adenosine resulting from circulating human CD39+ Treg and MDSCs, beyond stimulating the vascular endothelial cell proliferation, reduced markedly the migration of effector T cells into the TME, thus disabling the anti-tumor immunity [77,111].
An over-expression of A2B receptors was also observed in sections of neoplastic colorectal tissues in comparison with normal colonic tissues [112]. In the same study, the authors detected a marked presence of this receptor subtype in several human CRC-derived cell lines [112]. In this context, the treatment with selective A2B receptor antagonist, MRS1754, reduced the neoplastic growth of CRC cell lines, indicating a direct cancer-promoting property of this receptor subtype [112]. In this regard, other authors reported that also the A3 receptors, overexpressed in CRC tissues, can take a relevant part to CRC proliferation [113].
In summary, the extracellular adenosine, massively released in cancer tissue, participates actively to the generation of immunosuppressive and cancer growth-promoting microenvironment by regulating the functions of tumor cells, stromal cells, and tumor-infiltrating immune cells. In this regard, adenosine, through the engagement of its receptors expressed on immune cells, switches the phenotype of these cells from immune surveillance and host defense role towards a cancer-promoting phenotype, contributing to the generation of immunosuppressed niche. At the same time, beyond its involvement in the generation of an immune tolerant microenvironment suitable for tumor onset and progression, adenosine interferes also directly with cancer cells, spurring their proliferation, inhibiting the apoptotic process, promoting the angiogenic process, and predisposing the neoplastic cells to the metastatic dissemination.
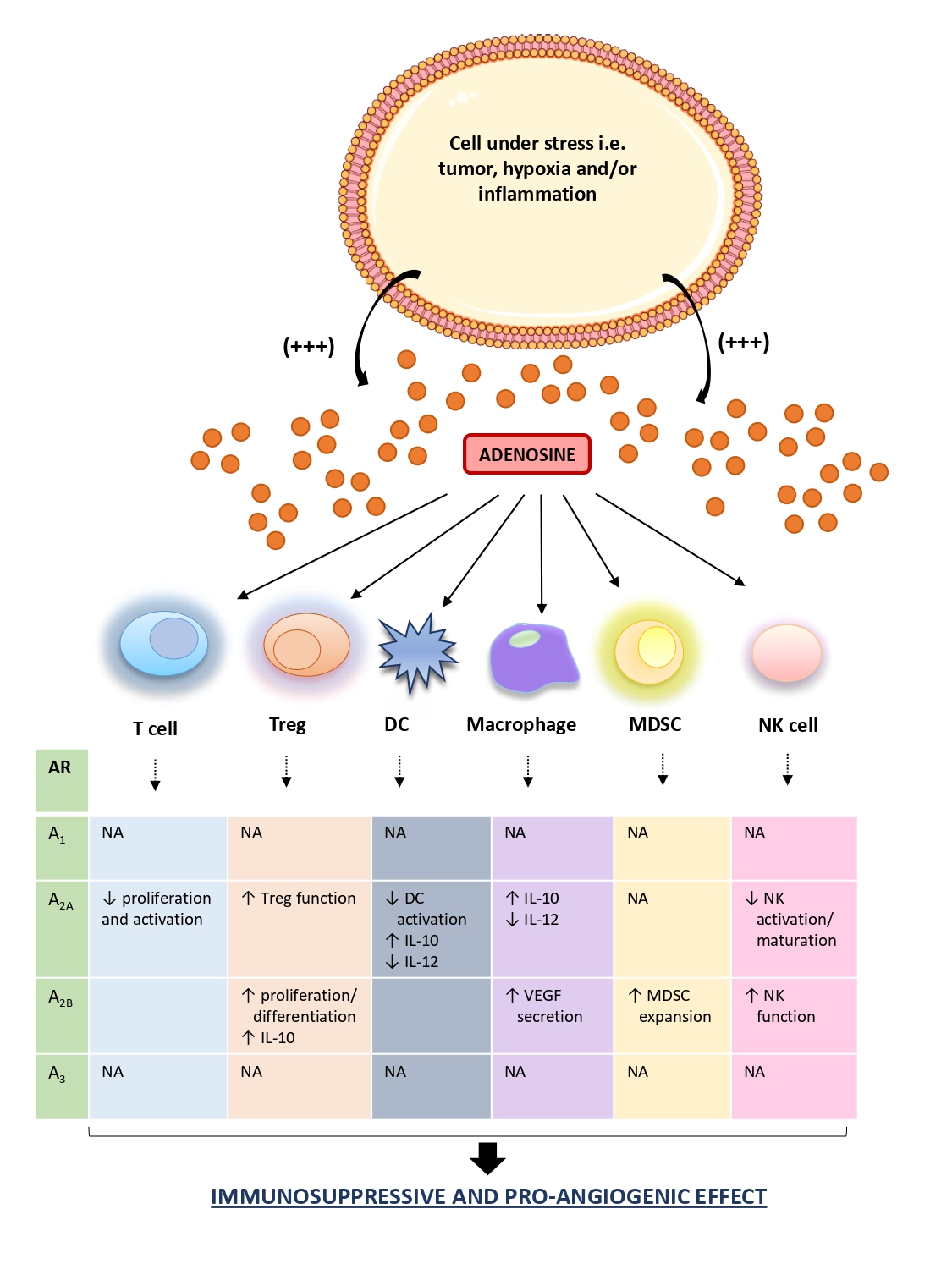
Figure 3. Schematic figure of adenosine and immune cell interactions in TME. In the tumor microenvironment, adenosine is markedly released in the extracellular space. In this context, it interacts with infiltrating immune cells (i.e., macrophages, T cell, DCs), triggering, and maintaining an immunosuppressed milieu, an ideal condition for the onset, and development of neoplasia. ↑: increase; ↓: decrease; AR: adenosine receptor; DC: dendritic cell; IL: interleukin; MDSC: myeloid-derived suppressor cell; NA: not available; NK: natural killer.
[1][2][3][4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82][83][84][85][86][87][88][89][90][91][92][93][94][95][96][97][98][99][100][101][102][103][104][105][106][107][108][109][110][111][112][113]
References
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Annals of African medicine 2019, 18, 121-126, doi:10.4103/aam.aam_56_18.
- Galdiero, M.R.; Marone, G.; Mantovani, A. Cancer Inflammation and Cytokines. Cold Spring Harbor perspectives in biology 2018, 10, doi:10.1101/cshperspect.a028662.
- Tuomisto, A.E.; Makinen, M.J.; Vayrynen, J.P. Systemic inflammation in colorectal cancer: Underlying factors, effects, and prognostic significance. World journal of gastroenterology 2019, 25, 4383-4404, doi:10.3748/wjg.v25.i31.4383.
- Beaugerie, L.; Svrcek, M.; Seksik, P.; Bouvier, A.M.; Simon, T.; Allez, M.; Brixi, H.; Gornet, J.M.; Altwegg, R.; Beau, P., et al. Risk of colorectal high-grade dysplasia and cancer in a prospective observational cohort of patients with inflammatory bowel disease. Gastroenterology 2013, 145, 166-175 e168, doi:10.1053/j.gastro.2013.03.044.
- Fan, Y.; Jin, X.; Man, C.; Gao, Z.; Wang, X. Meta-analysis of the association between the inflammatory potential of diet and colorectal cancer risk. Oncotarget 2017, 8, 59592-59600, doi:10.18632/oncotarget.19233.
- de Waal, G.M.; de Villiers, W.J.S.; Forgan, T.; Roberts, T.; Pretorius, E. Colorectal cancer is associated with increased circulating lipopolysaccharide, inflammation and hypercoagulability. Scientific reports 2020, 10, 8777, doi:10.1038/s41598-020-65324-2.
- Keller, D.S.; Windsor, A.; Cohen, R.; Chand, M. Colorectal cancer in inflammatory bowel disease: review of the evidence. Techniques in coloproctology 2019, 23, 3-13, doi:10.1007/s10151-019-1926-2.
- Lu, M.J.; Qiu, X.Y.; Mao, X.Q.; Li, X.T.; Zhang, H.J. Systematic review with meta-analysis: thiopurines decrease the risk of colorectal neoplasia in patients with inflammatory bowel disease. Alimentary pharmacology & therapeutics 2018, 47, 318-331, doi:10.1111/apt.14436.
- Zhou, Q.; Shen, Z.F.; Wu, B.S.; Xu, C.B.; He, Z.Q.; Chen, T.; Shang, H.T.; Xie, C.F.; Huang, S.Y.; Chen, Y.G., et al. Risk of Colorectal Cancer in Ulcerative Colitis Patients: A Systematic Review and Meta-Analysis. Gastroenterology research and practice 2019, 2019, 5363261, doi:10.1155/2019/5363261.
- D'Antongiovanni, V.; Martinelli, S.; Richter, S.; Canu, L.; Guasti, D.; Mello, T.; Romagnoli, P.; Pacak, K.; Eisenhofer, G.; Mannelli, M., et al. The microenvironment induces collective migration in SDHB-silenced mouse pheochromocytoma spheroids. Endocrine-related cancer 2017, 24, 555-564, doi:10.1530/ERC-17-0212.
- Richter, S.; D'Antongiovanni, V.; Martinelli, S.; Bechmann, N.; Riverso, M.; Poitz, D.M.; Pacak, K.; Eisenhofer, G.; Mannelli, M.; Rapizzi, E. Primary fibroblast co-culture stimulates growth and metabolism in Sdhb-impaired mouse pheochromocytoma MTT cells. Cell and tissue research 2018, 374, 473-485, doi:10.1007/s00441-018-2907-x.
- Li, W.; Zhang, X.; Wu, F.; Zhou, Y.; Bao, Z.; Li, H.; Zheng, P.; Zhao, S. Gastric cancer-derived mesenchymal stromal cells trigger M2 macrophage polarization that promotes metastasis and EMT in gastric cancer. Cell death & disease 2019, 10, 918, doi:10.1038/s41419-019-2131-y.
- Umakoshi, M.; Takahashi, S.; Itoh, G.; Kuriyama, S.; Sasaki, Y.; Yanagihara, K.; Yashiro, M.; Maeda, D.; Goto, A.; Tanaka, M. Macrophage-mediated transfer of cancer-derived components to stromal cells contributes to establishment of a pro-tumor microenvironment. Oncogene 2019, 38, 2162-2176, doi:10.1038/s41388-018-0564-x.
- Upadhyay, S.; Sharma, N.; Gupta, K.B.; Dhiman, M. Role of immune system in tumor progression and carcinogenesis. Journal of cellular biochemistry 2018, 119, 5028-5042, doi:10.1002/jcb.26663.
- Bonavita, E.; Galdiero, M.R.; Jaillon, S.; Mantovani, A. Phagocytes as Corrupted Policemen in Cancer-Related Inflammation. Advances in cancer research 2015, 128, 141-171, doi:10.1016/bs.acr.2015.04.013.
- Nakatsumi, H.; Matsumoto, M.; Nakayama, K.I. Noncanonical Pathway for Regulation of CCL2 Expression by an mTORC1-FOXK1 Axis Promotes Recruitment of Tumor-Associated Macrophages. Cell reports 2017, 21, 2471-2486, doi:10.1016/j.celrep.2017.11.014.
- Seager, R.J.; Hajal, C.; Spill, F.; Kamm, R.D.; Zaman, M.H. Dynamic interplay between tumour, stroma and immune system can drive or prevent tumour progression. Convergent science physical oncology 2017, 3, doi:10.1088/2057-1739/aa7e86.
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nature reviews. Immunology 2017, 17, 559-572, doi:10.1038/nri.2017.49.
- Schetter, A.J.; Heegaard, N.H.; Harris, C.C. Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis 2010, 31, 37-49, doi:10.1093/carcin/bgp272.
- Blay, J.; White, T.D.; Hoskin, D.W. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer research 1997, 57, 2602-2605.
- Arab, S.; Hadjati, J. Adenosine Blockage in Tumor Microenvironment and Improvement of Cancer Immunotherapy. Immune network 2019, 19, e23, doi:10.4110/in.2019.19.e23.
- Hoskin, D.W.; Butler, J.J.; Drapeau, D.; Haeryfar, S.M.; Blay, J. Adenosine acts through an A3 receptor to prevent the induction of murine anti-CD3-activated killer T cells. International journal of cancer 2002, 99, 386-395, doi:10.1002/ijc.10325.
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Hasko, G. Immunity, inflammation and cancer: a leading role for adenosine. Nature reviews. Cancer 2013, 13, 842-857, doi:10.1038/nrc3613.
- Antonioli, L.; Novitskiy, S.V.; Sachsenmeier, K.F.; Fornai, M.; Blandizzi, C.; Hasko, G. Switching off CD73: a way to boost the activity of conventional and targeted antineoplastic therapies. Drug discovery today 2017, 22, 1686-1696, doi:10.1016/j.drudis.2017.06.005.
- Liu, H.; Xia, Y. Beneficial and detrimental role of adenosine signaling in diseases and therapy. Journal of applied physiology 2015, 119, 1173-1182, doi:10.1152/japplphysiol.00350.2015.
- Lofgren, L.; Pehrsson, S.; Hagglund, G.; Tjellstrom, H.; Nylander, S. Accurate measurement of endogenous adenosine in human blood. PloS one 2018, 13, e0205707, doi:10.1371/journal.pone.0205707.
- Deussen, A.; Stappert, M.; Schafer, S.; Kelm, M. Quantification of extracellular and intracellular adenosine production: understanding the transmembranous concentration gradient. Circulation 1999, 99, 2041-2047, doi:10.1161/01.cir.99.15.2041.
- Hasko, G.; Cronstein, B.N. Adenosine: an endogenous regulator of innate immunity. Trends in immunology 2004, 25, 33-39, doi:10.1016/j.it.2003.11.003.
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Hasko, G. The Purinergic System as a Pharmacological Target for the Treatment of Immune-Mediated Inflammatory Diseases. Pharmacological reviews 2019, 71, 345-382, doi:10.1124/pr.117.014878.
- Sitkovsky, M.V.; Kjaergaard, J.; Lukashev, D.; Ohta, A. Hypoxia-adenosinergic immunosuppression: tumor protection by T regulatory cells and cancerous tissue hypoxia. Clinical cancer research : an official journal of the American Association for Cancer Research 2008, 14, 5947-5952, doi:10.1158/1078-0432.CCR-08-0229.
- Spychala, J. Tumor-promoting functions of adenosine. Pharmacology & therapeutics 2000, 87, 161-173, doi:10.1016/s0163-7258(00)00053-x.
- Hasko, G.; Pacher, P. Regulation of macrophage function by adenosine. Arteriosclerosis, thrombosis, and vascular biology 2012, 32, 865-869, doi:10.1161/ATVBAHA.111.226852.
- Linden, J. Adenosine metabolism and cancer. Focus on "Adenosine downregulates DPPIV on HT-29 colon cancer cells by stimulating protein tyrosine phosphatases and reducing ERK1/2 activity via a novel pathway". American journal of physiology. Cell physiology 2006, 291, C405-406, doi:10.1152/ajpcell.00242.2006.
- Ma, Y.; Zhang, J.; Zhang, Q.; Chen, P.; Song, J.; Yu, S.; Liu, H.; Liu, F.; Song, C.; Yang, D., et al. Adenosine induces apoptosis in human liver cancer cells through ROS production and mitochondrial dysfunction. Biochemical and biophysical research communications 2014, 448, 8-14, doi:10.1016/j.bbrc.2014.04.007.
- Antonioli, L.; Colucci, R.; La Motta, C.; Tuccori, M.; Awwad, O.; Da Settimo, F.; Blandizzi, C.; Fornai, M. Adenosine deaminase in the modulation of immune system and its potential as a novel target for treatment of inflammatory disorders. Current drug targets 2012, 13, 842-862, doi:10.2174/138945012800564095.
- Eltzschig, H.K. Extracellular adenosine signaling in molecular medicine. Journal of molecular medicine 2013, 91, 141-146, doi:10.1007/s00109-013-0999-z.
- Pastor-Anglada, M.; Urtasun, N.; Perez-Torras, S. Intestinal Nucleoside Transporters: Function, Expression, and Regulation. Comprehensive Physiology 2018, 8, 1003-1017, doi:10.1002/cphy.c170039.
- Antonioli, L.; Fornai, M.; Colucci, R.; Ghisu, N.; Tuccori, M.; Del Tacca, M.; Blandizzi, C. Regulation of enteric functions by adenosine: pathophysiological and pharmacological implications. Pharmacology & therapeutics 2008, 120, 233-253, doi:10.1016/j.pharmthera.2008.08.010.
- Archer, R.G.; Pitelka, V.; Hammond, J.R. Nucleoside transporter subtype expression and function in rat skeletal muscle microvascular endothelial cells. British journal of pharmacology 2004, 143, 202-214, doi:10.1038/sj.bjp.0705921.
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pathological overproduction: the bad side of adenosine. British journal of pharmacology 2017, 174, 1945-1960, doi:10.1111/bph.13763.
- Chen, S.; Wainwright, D.A.; Wu, J.D.; Wan, Y.; Matei, D.E.; Zhang, Y.; Zhang, B. CD73: an emerging checkpoint for cancer immunotherapy. Immunotherapy 2019, 11, 983-997, doi:10.2217/imt-2018-0200.
- Yang, D.; Yaguchi, T.; Lim, C.R.; Ishizawa, Y.; Nakano, T.; Nishizaki, T. Tuning of apoptosis-mediator gene transcription in HepG2 human hepatoma cells through an adenosine signal. Cancer letters 2010, 291, 225-229, doi:10.1016/j.canlet.2009.10.016.
- Sheth, S.; Brito, R.; Mukherjea, D.; Rybak, L.P.; Ramkumar, V. Adenosine receptors: expression, function and regulation. International journal of molecular sciences 2014, 15, 2024-2052, doi:10.3390/ijms15022024.
- Hoskin, D.W.; Mader, J.S.; Furlong, S.J.; Conrad, D.M.; Blay, J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review). International journal of oncology 2008, 32, 527-535.
- Klinger, M.; Freissmuth, M.; Nanoff, C. Adenosine receptors: G protein-mediated signalling and the role of accessory proteins. Cellular signalling 2002, 14, 99-108, doi:10.1016/s0898-6568(01)00235-2.
- da Silva, C.G.; Jarzyna, R.; Specht, A.; Kaczmarek, E. Extracellular nucleotides and adenosine independently activate AMP-activated protein kinase in endothelial cells: involvement of P2 receptors and adenosine transporters. Circulation research 2006, 98, e39-47, doi:10.1161/01.RES.0000215436.92414.1d.
- Boison, D. Adenosine kinase: exploitation for therapeutic gain. Pharmacological reviews 2013, 65, 906-943, doi:10.1124/pr.112.006361.
- Mato, J.M.; Martinez-Chantar, M.L.; Lu, S.C. Methionine metabolism and liver disease. Annual review of nutrition 2008, 28, 273-293, doi:10.1146/annurev.nutr.28.061807.155438.
- Nadeem, M.S.; Kumar, V.; Al-Abbasi, F.A.; Kamal, M.A.; Anwar, F. Risk of colorectal cancer in inflammatory bowel diseases. Seminars in cancer biology 2019, 10.1016/j.semcancer.2019.05.001, doi:10.1016/j.semcancer.2019.05.001.
- Andersen, N.N.; Jess, T. Has the risk of colorectal cancer in inflammatory bowel disease decreased? World journal of gastroenterology 2013, 19, 7561-7568, doi:10.3748/wjg.v19.i43.7561.
- Molodecky, N.A.; Kareemi, H.; Parab, R.; Barkema, H.W.; Quan, H.; Myers, R.P.; Kaplan, G.G. Incidence of primary sclerosing cholangitis: a systematic review and meta-analysis. Hepatology 2011, 53, 1590-1599, doi:10.1002/hep.24247.
- Sebastian, S.; Hernandez, V.; Myrelid, P.; Kariv, R.; Tsianos, E.; Toruner, M.; Marti-Gallostra, M.; Spinelli, A.; van der Meulen-de Jong, A.E.; Yuksel, E.S., et al. Colorectal cancer in inflammatory bowel disease: results of the 3rd ECCO pathogenesis scientific workshop (I). Journal of Crohn's & colitis 2014, 8, 5-18, doi:10.1016/j.crohns.2013.04.008.
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Current protocols in immunology 2014, 104, 15 25 11-15 25 14, doi:10.1002/0471142735.im1525s104.
- Alex, P.; Zachos, N.C.; Nguyen, T.; Gonzales, L.; Chen, T.E.; Conklin, L.S.; Centola, M.; Li, X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflammatory bowel diseases 2009, 15, 341-352, doi:10.1002/ibd.20753.
- Linden, J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annual review of pharmacology and toxicology 2001, 41, 775-787, doi:10.1146/annurev.pharmtox.41.1.775.
- Hasko, G.; Cronstein, B. Regulation of inflammation by adenosine. Frontiers in immunology 2013, 4, 85, doi:10.3389/fimmu.2013.00085.
- Siegmund, B.; Rieder, F.; Albrich, S.; Wolf, K.; Bidlingmaier, C.; Firestein, G.S.; Boyle, D.; Lehr, H.A.; Loher, F.; Hartmann, G., et al. Adenosine kinase inhibitor GP515 improves experimental colitis in mice. The Journal of pharmacology and experimental therapeutics 2001, 296, 99-105.
- Pallio, G.; Bitto, A.; Pizzino, G.; Galfo, F.; Irrera, N.; Squadrito, F.; Squadrito, G.; Pallio, S.; Anastasi, G.P.; Cutroneo, G., et al. Adenosine Receptor Stimulation by Polydeoxyribonucleotide Improves Tissue Repair and Symptomology in Experimental Colitis. Frontiers in pharmacology 2016, 7, 273, doi:10.3389/fphar.2016.00273.
- Selmeczy, Z.; Csoka, B.; Pacher, P.; Vizi, E.S.; Hasko, G. The adenosine A2A receptor agonist CGS 21680 fails to ameliorate the course of dextran sulphate-induced colitis in mice. Inflammation research : official journal of the European Histamine Research Society ... [et al.] 2007, 56, 204-209, doi:10.1007/s00011-006-6150-7.
- Kolachala, V.; Asamoah, V.; Wang, L.; Obertone, T.S.; Ziegler, T.R.; Merlin, D.; Sitaraman, S.V. TNF-alpha upregulates adenosine 2b (A2b) receptor expression and signaling in intestinal epithelial cells: a basis for A2bR overexpression in colitis. Cellular and molecular life sciences : CMLS 2005, 62, 2647-2657, doi:10.1007/s00018-005-5328-4.
- Kolachala, V.; Ruble, B.; Vijay-Kumar, M.; Wang, L.; Mwangi, S.; Figler, H.; Figler, R.; Srinivasan, S.; Gewirtz, A.; Linden, J., et al. Blockade of adenosine A2B receptors ameliorates murine colitis. British journal of pharmacology 2008, 155, 127-137, doi:10.1038/bjp.2008.227.
- Frick, J.S.; MacManus, C.F.; Scully, M.; Glover, L.E.; Eltzschig, H.K.; Colgan, S.P. Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. Journal of immunology 2009, 182, 4957-4964, doi:10.4049/jimmunol.0801324.
- Aherne, C.M.; Saeedi, B.; Collins, C.B.; Masterson, J.C.; McNamee, E.N.; Perrenoud, L.; Rapp, C.R.; Curtis, V.F.; Bayless, A.; Fletcher, A., et al. Epithelial-specific A2B adenosine receptor signaling protects the colonic epithelial barrier during acute colitis. Mucosal immunology 2015, 8, 1324-1338, doi:10.1038/mi.2015.22.
- Ren, T.; Grants, I.; Alhaj, M.; McKiernan, M.; Jacobson, M.; Hassanain, H.H.; Frankel, W.; Wunderlich, J.; Christofi, F.L. Impact of disrupting adenosine A(3) receptors (A(3)(-)/(-) AR) on colonic motility or progression of colitis in the mouse. Inflammatory bowel diseases 2011, 17, 1698-1713, doi:10.1002/ibd.21553.
- Mabley, J.; Soriano, F.; Pacher, P.; Hasko, G.; Marton, A.; Wallace, R.; Salzman, A.; Szabo, C. The adenosine A3 receptor agonist, N6-(3-iodobenzyl)-adenosine-5'-N-methyluronamide, is protective in two murine models of colitis. European journal of pharmacology 2003, 466, 323-329, doi:10.1016/s0014-2999(03)01570-x.
- Ren, T.; Tian, T.; Feng, X.; Ye, S.; Wang, H.; Wu, W.; Qiu, Y.; Yu, C.; He, Y.; Zeng, J., et al. An adenosine A3 receptor agonist inhibits DSS-induced colitis in mice through modulation of the NF-kappaB signaling pathway. Scientific reports 2015, 5, 9047, doi:10.1038/srep09047.
- Wu, W.; He, Y.; Feng, X.; Ye, S.; Wang, H.; Tan, W.; Yu, C.; Hu, J.; Zheng, R.; Zhou, Y. MicroRNA-206 is involved in the pathogenesis of ulcerative colitis via regulation of adenosine A3 receptor. Oncotarget 2017, 8, 705-721, doi:10.18632/oncotarget.13525.
- Ren, T.; Qiu, Y.; Wu, W.; Feng, X.; Ye, S.; Wang, Z.; Tian, T.; He, Y.; Yu, C.; Zhou, Y. Activation of adenosine A3 receptor alleviates TNF-alpha-induced inflammation through inhibition of the NF-kappaB signaling pathway in human colonic epithelial cells. Mediators of inflammation 2014, 2014, 818251, doi:10.1155/2014/818251.
- Tian, T.; Zhou, Y.; Feng, X.; Ye, S.; Wang, H.; Wu, W.; Tan, W.; Yu, C.; Hu, J.; Zheng, R., et al. MicroRNA-16 is putatively involved in the NF-kappaB pathway regulation in ulcerative colitis through adenosine A2a receptor (A2aAR) mRNA targeting. Scientific reports 2016, 6, 30824, doi:10.1038/srep30824.
- Ren, T.H.; Lv, M.M.; An, X.M.; Leung, W.K.; Seto, W.K. Activation of adenosine A3 receptor inhibits inflammatory cytokine production in colonic mucosa of patients with ulcerative colitis by down-regulating the nuclear factor-kappa B signaling. Journal of digestive diseases 2020, 21, 38-45, doi:10.1111/1751-2980.12831.
- Danese, S.; Malesci, A.; Vetrano, S. Colitis-associated cancer: the dark side of inflammatory bowel disease. Gut 2011, 60, 1609-1610, doi:10.1136/gutjnl-2011-300953.
- Terzic, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101-2114 e2105, doi:10.1053/j.gastro.2010.01.058.
- Parang, B.; Barrett, C.W.; Williams, C.S. AOM/DSS Model of Colitis-Associated Cancer. Methods in molecular biology 2016, 1422, 297-307, doi:10.1007/978-1-4939-3603-8_26.
- Aghaei, M.; Karami-Tehrani, F.; Panjehpour, M.; Salami, S.; Fallahian, F. Adenosine induces cell-cycle arrest and apoptosis in androgen-dependent and -independent prostate cancer cell lines, LNcap-FGC-10, DU-145, and PC3. The Prostate 2012, 72, 361-375, doi:10.1002/pros.21438.
- Yang, D.; Yaguchi, T.; Yamamoto, H.; Nishizaki, T. Intracellularly transported adenosine induces apoptosis in HuH-7 human hepatoma cells by downregulating c-FLIP expression causing caspase-3/-8 activation. Biochemical pharmacology 2007, 73, 1665-1675, doi:10.1016/j.bcp.2007.01.020.
- Wu, X.R.; He, X.S.; Chen, Y.F.; Yuan, R.X.; Zeng, Y.; Lian, L.; Zou, Y.F.; Lan, N.; Wu, X.J.; Lan, P. High expression of CD73 as a poor prognostic biomarker in human colorectal cancer. Journal of surgical oncology 2012, 106, 130-137, doi:10.1002/jso.23056.
- Limagne, E.; Euvrard, R.; Thibaudin, M.; Rebe, C.; Derangere, V.; Chevriaux, A.; Boidot, R.; Vegran, F.; Bonnefoy, N.; Vincent, J., et al. Accumulation of MDSC and Th17 Cells in Patients with Metastatic Colorectal Cancer Predicts the Efficacy of a FOLFOX-Bevacizumab Drug Treatment Regimen. Cancer research 2016, 76, 5241-5252, doi:10.1158/0008-5472.CAN-15-3164.
- Ahlmanner, F.; Sundstrom, P.; Akeus, P.; Eklof, J.; Borjesson, L.; Gustavsson, B.; Lindskog, E.B.; Raghavan, S.; Quiding-Jarbrink, M. CD39(+) regulatory T cells accumulate in colon adenocarcinomas and display markers of increased suppressive function. Oncotarget 2018, 9, 36993-37007, doi:10.18632/oncotarget.26435.
- Zhang, B.; Cheng, B.; Li, F.S.; Ding, J.H.; Feng, Y.Y.; Zhuo, G.Z.; Wei, H.F.; Zhao, K. High expression of CD39/ENTPD1 in malignant epithelial cells of human rectal adenocarcinoma. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine 2015, 36, 9411-9419, doi:10.1007/s13277-015-3683-9.
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The adenosine pathway in immuno-oncology. Nature reviews. Clinical oncology 2020, 10.1038/s41571-020-0382-2, doi:10.1038/s41571-020-0382-2.
- Bowser, J.L.; Phan, L.H.; Eltzschig, H.K. The Hypoxia-Adenosine Link during Intestinal Inflammation. Journal of immunology 2018, 200, 897-907, doi:10.4049/jimmunol.1701414.
- Sitkovsky, M.; Lukashev, D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nature reviews. Immunology 2005, 5, 712-721, doi:10.1038/nri1685.
- Lukashev, D.; Klebanov, B.; Kojima, H.; Grinberg, A.; Ohta, A.; Berenfeld, L.; Wenger, R.H.; Ohta, A.; Sitkovsky, M. Cutting edge: hypoxia-inducible factor 1alpha and its activation-inducible short isoform I.1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. Journal of immunology 2006, 177, 4962-4965, doi:10.4049/jimmunol.177.8.4962.
- Lukashev, D.; Ohta, A.; Sitkovsky, M. Hypoxia-dependent anti-inflammatory pathways in protection of cancerous tissues. Cancer metastasis reviews 2007, 26, 273-279, doi:10.1007/s10555-007-9054-2.
- Wu, R.; Chen, Y.; Li, F.; Li, W.; Zhou, H.; Yang, Y.; Pei, Z. Effects of CD73 on human colorectal cancer cell growth in vivo and in vitro. Oncology reports 2016, 35, 1750-1756, doi:10.3892/or.2015.4512.
- Yu, M.; Guo, G.; Huang, L.; Deng, L.; Chang, C.S.; Achyut, B.R.; Canning, M.; Xu, N.; Arbab, A.S.; Bollag, R.J., et al. CD73 on cancer-associated fibroblasts enhanced by the A2B-mediated feedforward circuit enforces an immune checkpoint. Nature communications 2020, 11, 515, doi:10.1038/s41467-019-14060-x.
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F., et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-beta-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017, 6, e1320011, doi:10.1080/2162402X.2017.1320011.
- Richard, E.; Alam, S.M.; Arredondo-Vega, F.X.; Patel, D.D.; Hershfield, M.S. Clustered charged amino acids of human adenosine deaminase comprise a functional epitope for binding the adenosine deaminase complexing protein CD26/dipeptidyl peptidase IV. The Journal of biological chemistry 2002, 277, 19720-19726, doi:10.1074/jbc.M111901200.
- Tan, E.Y.; Mujoomdar, M.; Blay, J. Adenosine down-regulates the surface expression of dipeptidyl peptidase IV on HT-29 human colorectal carcinoma cells: implications for cancer cell behavior. The American journal of pathology 2004, 165, 319-330, doi:10.1016/S0002-9440(10)63299-3.
- Tan, E.Y.; Richard, C.L.; Zhang, H.; Hoskin, D.W.; Blay, J. Adenosine downregulates DPPIV on HT-29 colon cancer cells by stimulating protein tyrosine phosphatase(s) and reducing ERK1/2 activity via a novel pathway. American journal of physiology. Cell physiology 2006, 291, C433-444, doi:10.1152/ajpcell.00238.2005.
- Decking, U.K.; Schlieper, G.; Kroll, K.; Schrader, J. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circulation research 1997, 81, 154-164, doi:10.1161/01.res.81.2.154.
- Ohta, A. A Metabolic Immune Checkpoint: Adenosine in Tumor Microenvironment. Frontiers in immunology 2016, 7, 109, doi:10.3389/fimmu.2016.00109.
- Sitkovsky, M.V.; Hatfield, S.; Abbott, R.; Belikoff, B.; Lukashev, D.; Ohta, A. Hostile, hypoxia-A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer immunology research 2014, 2, 598-605, doi:10.1158/2326-6066.CIR-14-0075.
- Sun, X.; Wu, Y.; Gao, W.; Enjyoji, K.; Csizmadia, E.; Muller, C.E.; Murakami, T.; Robson, S.C. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology 2010, 139, 1030-1040, doi:10.1053/j.gastro.2010.05.007.
- Iannone, R.; Miele, L.; Maiolino, P.; Pinto, A.; Morello, S. Blockade of A2b adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse model of melanoma. Neoplasia 2013, 15, 1400-1409.
- Novitskiy, S.V.; Ryzhov, S.; Zaynagetdinov, R.; Goldstein, A.E.; Huang, Y.; Tikhomirov, O.Y.; Blackburn, M.R.; Biaggioni, I.; Carbone, D.P.; Feoktistov, I., et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 2008, 112, 1822-1831, doi:10.1182/blood-2008-02-136325.
- Romio, M.; Reinbeck, B.; Bongardt, S.; Huls, S.; Burghoff, S.; Schrader, J. Extracellular purine metabolism and signaling of CD73-derived adenosine in murine Treg and Teff cells. American journal of physiology. Cell physiology 2011, 301, C530-539, doi:10.1152/ajpcell.00385.2010.
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Frontiers in immunology 2012, 3, 190, doi:10.3389/fimmu.2012.00190.
- Cekic, C.; Day, Y.J.; Sag, D.; Linden, J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer research 2014, 74, 7250-7259, doi:10.1158/0008-5472.CAN-13-3583.
- Beavis, P.A.; Divisekera, U.; Paget, C.; Chow, M.T.; John, L.B.; Devaud, C.; Dwyer, K.; Stagg, J.; Smyth, M.J.; Darcy, P.K. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proceedings of the National Academy of Sciences of the United States of America 2013, 110, 14711-14716, doi:10.1073/pnas.1308209110.
- Panther, E.; Corinti, S.; Idzko, M.; Herouy, Y.; Napp, M.; la Sala, A.; Girolomoni, G.; Norgauer, J. Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the T-cell stimulatory capacity of human dendritic cells. Blood 2003, 101, 3985-3990, doi:10.1182/blood-2002-07-2113.
- Fukata, M.; Shang, L.; Santaolalla, R.; Sotolongo, J.; Pastorini, C.; Espana, C.; Ungaro, R.; Harpaz, N.; Cooper, H.S.; Elson, G., et al. Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis-associated tumorigenesis. Inflammatory bowel diseases 2011, 17, 1464-1473, doi:10.1002/ibd.21527.
- Francescone, R.; Hou, V.; Grivennikov, S.I. Cytokines, IBD, and colitis-associated cancer. Inflammatory bowel diseases 2015, 21, 409-418, doi:10.1097/MIB.0000000000000236.
- Ryzhov, S.; Novitskiy, S.V.; Goldstein, A.E.; Biktasova, A.; Blackburn, M.R.; Biaggioni, I.; Dikov, M.M.; Feoktistov, I. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. Journal of immunology 2011, 187, 6120-6129, doi:10.4049/jimmunol.1101225.
- Ehrentraut, H.; Westrich, J.A.; Eltzschig, H.K.; Clambey, E.T. Adora2b adenosine receptor engagement enhances regulatory T cell abundance during endotoxin-induced pulmonary inflammation. PloS one 2012, 7, e32416, doi:10.1371/journal.pone.0032416.
- Sorrentino, C.; Miele, L.; Porta, A.; Pinto, A.; Morello, S. Myeloid-derived suppressor cells contribute to A2B adenosine receptor-induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget 2015, 6, 27478-27489, doi:10.18632/oncotarget.4393.
- Wang, L.; Fan, J.; Thompson, L.F.; Zhang, Y.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. The Journal of clinical investigation 2011, 121, 2371-2382, doi:10.1172/JCI45559.
- Ryzhov, S.V.; Pickup, M.W.; Chytil, A.; Gorska, A.E.; Zhang, Q.; Owens, P.; Feoktistov, I.; Moses, H.L.; Novitskiy, S.V. Role of TGF-beta signaling in generation of CD39+CD73+ myeloid cells in tumors. Journal of immunology 2014, 193, 3155-3164, doi:10.4049/jimmunol.1400578.
- Harish, A.; Hohana, G.; Fishman, P.; Arnon, O.; Bar-Yehuda, S. A3 adenosine receptor agonist potentiates natural killer cell activity. International journal of oncology 2003, 23, 1245-1249.
- Wang, X.; Chen, D. Purinergic Regulation of Neutrophil Function. Frontiers in immunology 2018, 9, 399, doi:10.3389/fimmu.2018.00399.
- Sundstrom, P.; Stenstad, H.; Langenes, V.; Ahlmanner, F.; Theander, L.; Ndah, T.G.; Fredin, K.; Borjesson, L.; Gustavsson, B.; Bastid, J., et al. Regulatory T Cells from Colon Cancer Patients Inhibit Effector T-cell Migration through an Adenosine-Dependent Mechanism. Cancer immunology research 2016, 4, 183-193, doi:10.1158/2326-6066.CIR-15-0050.
- Ma, D.F.; Kondo, T.; Nakazawa, T.; Niu, D.F.; Mochizuki, K.; Kawasaki, T.; Yamane, T.; Katoh, R. Hypoxia-inducible adenosine A2B receptor modulates proliferation of colon carcinoma cells. Human pathology 2010, 41, 1550-1557, doi:10.1016/j.humpath.2010.04.008.
- Gessi, S.; Cattabriga, E.; Avitabile, A.; Gafa, R.; Lanza, G.; Cavazzini, L.; Bianchi, N.; Gambari, R.; Feo, C.; Liboni, A., et al. Elevated expression of A3 adenosine receptors in human colorectal cancer is reflected in peripheral blood cells. Clinical cancer research : an official journal of the American Association for Cancer Research 2004, 10, 5895-5901, doi:10.1158/1078-0432.CCR-1134-03.


