 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Herminia González-Navarro | + 2265 word(s) | 2265 | 2020-07-16 11:36:30 | | | |
| 2 | Nicole Yin | -247 word(s) | 2018 | 2020-07-22 04:01:42 | | | | |
| 3 | Nicole Yin | -9 word(s) | 2009 | 2020-11-05 09:00:24 | | |
Video Upload Options
Cholesterol, the most important sterol in mammals, helps maintain plasma membrane fluidity and is a precursor of bile acids, oxysterols, and steroid hormones. Cholesterol in the body is obtained from the diet or can be de novo synthetized. Cholesterol homeostasis is mainly regulated by the liver, where cholesterol is packed in lipoproteins for transport through a tightly regulated process. Changes in circulating lipoprotein cholesterol levels lead to atherosclerosis development, which is initiated by an accumulation of modified lipoproteins in the subendothelial space; this induces significant changes in immune cell differentiation and function. In this entry, we describe the main regulatory pathways and mechanisms of cholesterol metabolism.
1. Definition
Cholesterol is the main sterol in mammals and has a key role in the plasma membrane where it is responsible for modulating membrane fluidity, permeability, and signaling[1].
2. Introduction
It is also found in the endoplasmic reticulum (ER) membrane in small amounts where it is essential for its metabolic regulation[2]. Cholesterol is also involved in cellular proliferation and embryonic signaling and is a precursor of bile acids, oxysterols, and all steroid hormones.
Cholesterol blood levels are determined by the ingestion of dietary cholesterol and by de novo synthetized cholesterol from acetyl coenzyme A (acetyl-CoA). Cholesterol homeostasis is tightly regulated by the liver, which controls lipoprotein assembling, secretion, and catabolism[1]. Altered cholesterol metabolism and regulation of its circulating levels have a major impact on atherosclerosis, a chronic inflammatory disease. Given that cholesterol is needed for all cellular membranes, genes involved in cholesterol biosynthesis, regulatory pathways, transport, and receptors are ubiquitous across all cell types.
3. Cholesterol Metabolism
In the sections below, we provide an overview of the main and most current pathways and key modulators of intracellular cholesterol metabolism and circulating cholesterol levels, which are also summarized in Figures 1 and 2.
3.1. Cholesterol Absorption and Metabolism
In the intestinal lumen, cholesterol binds to bile salt micelles and is transported by Niemann–Pick C1-Like 1 (NPC1L1) through the enterocyte brush border membrane via clathrin-mediated endocytosis[1][2][3] (Figure 1). NPC1L1 is a cholesterol-sensing receptor that traffics from the endocytic recycling compartment to the membrane[3]. During this process, ras-associated binding protein (RAB)11a bound to NPC1L1 is replaced by cell division control protein (CDC)42, which activates actin polymerization promoters that traffic NPC1L1 back to the cell surface. Under cholesterol depletion condition, NPC1L1 is also regulated by sterol regulatory element-binding protein 2 (SREBP2) transcription factor and lysosomal and ubiquitin–proteasome degradation[3][4]. Inside the enterocytes, the cholesterol is re-esterified by acetyl-CoA (Ac-CoA) acetyltransferase 2 (ACAT2) enzyme[5] and assembled into chylomicrons, a triglyceride-rich lipoprotein type with a cholesteryl ester-rich core and a single molecule of apolipoprotein B (ApoB)48 protein on its surface. These nascent chylomicrons are secreted into the intercellular space, migrate to the lamina propria, and enter the lacteal vessels of the lymphatic system for transport through the portal vein into the liver or into the circulation via the thoracic duct[2](Figure 1).
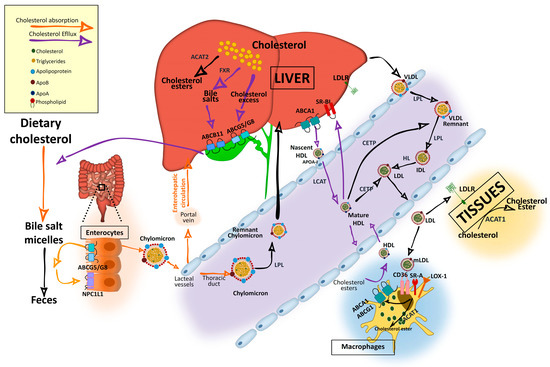
Figure 1. Cholesterol and lipoprotein metabolism. Dietary cholesterol is digested, solubilized in bile salt micelles in the intestine, and absorbed at the apical region of enterocytes by NPC1L1, where it is assembled in nascent chylomicrons. Chylomicrons will be transported to the liver through the portal vein of the enterohepatic circulation although they can enter into the circulation through the thoracic duct. Cholesterol can also be synthetized endogenously from Ac-CoA. In the liver, it is assembled in very low density lipoproteins (VLDLs), which are secreted into the circulation. Triglycerides in chylomicrons and VLDL are hydrolyzed by lipoprotein lipase (LPL), increasing their density and generating chylomicron remnants and low-density lipoprotein (LDL) particles that can be further catabolized by hepatic lipase (HL). VLDL and LDL are also enriched in cholesterol by cholesteryl ester transfer protein (CETP), which transfers cholesteryl esters from high-density lipoprotein (HDL). Resultant chylomicron remnants are taken up by the liver and the components enter into the lipoprotein packaging. LDL delivers cholesterol by an LDL receptor (LDLr)-mediated endocytic process to most of the tissues. In the subendothelial space, LDL undergoes a variety of modifications, such as oxidation, acetylation, or aggregation to generate modified LDL (mLDL), which is taken up by macrophages through scavenger receptor (SR)-A, cluster of differentiation (CD)36, and low-density lipoprotein receptor-1 (LOX-1) to become foam cells. The excess of cholesterol undergo an efflux process mediated by adenosine triphosphate-binding cassette transporter A1 (ABCA1) and adenosine triphosphate-binding cassette transporter G1 (ABCG1), which interact with apoliproprotein A (ApoA)-I of the cholesterol acceptor nascent discoidal lipid-poor HDL. Mature spherical lipid-rich HDL returns by the reverse cholesterol transport to the liver where it delivers cholesterol through scavenger receptor class B type 1 (SRBI). The liver stores cholesterol that can be used to synthetize bile salts in a farnesoid X receptor (FXR)-regulated pathway with the rate-limiting enzyme cytochrome P450 family 7 subfamily A member 1 (CYP7A1). Cholesterol can also be secreted into the gallbladder by adenosine triphosphate-binding cassette transporter G5/G8 (ABCG5/G8) along with bile salts, which are secreted by adenosine triphosphate-binding cassette subfamily B member 11 (ABCB11) until they are secreted in the intestine for elimination with the feces. Reabsorption by NPC1L1 at the apical side of enterocytes can incorporate cholesterol back into chylomicrons. For intracellular storage, cholesterol is esterified by ACAT2 in hepatocytes and by ACAT1 in the rest of the cell types.
3.2. Endogenous Synthesis of Cholesterol
Cellular depletion of cholesterol activates endogenous cholesterol biosynthesis from Ac-CoA molecules, in an energetically expensive anabolic pathway regulated by SREBP2 and the 3-hydroxy-3-methyl-glutaryl coenzyme A reductase (HMG-CoAR). SREBP2 is synthetized as an ER-anchored precursor that is translocated with the SREBP cleavage activating protein (SCAP) to the Golgi apparatus for activation through cleavage by site 1 protease (SP1) and SP2. Insulin-induced gene 1 (INSIG1) and INSIG2 proteins retain SREBP–SCAP dimer interaction when SCAP sterol-sensing domains (SSD) detect ER membrane cholesterol over 5 mol % of total lipids[3][4][6][7](Figure 2).
Activated nuclear SREBP binds to sterol regulatory element (SRE) sequences to activate transcription in target genes including its own; its synergistic inductors nuclear transcription factor Y subunit alpha (NF-Y) and SP1; its inhibitor FOXO3; and HMG-CoAR, an ER-glycoprotein that converts the HMG-CoA to mevalonate and is inhibited by non-sterol isoprenoids[3] [3]. Cholesterol abundance diminishes HMG-CoAR activity by INSIG1 and INSIG2, which bind to HMG-CoAR with ubiquitin ligases for degradation through different pathways[8](Figure 2). Oxysterols; methylated sterols; geranylgeraniol, an intermediate of vitamin biosynthesis; and two vitamin family members (δ- and γ-tocotrienol) are also strong inducers of HMG-CoAR degradation. HMG-CoAR can reversibly be inhibited by phosphorylation[3][4](Figure 2).
Hepatic de novo synthesized cholesterol is assembled in the Golgi apparatus in very low density lipoprotein (VLDL) particles, which are triglyceride-rich and cholesteryl ester-poor lipoproteins with a single molecule of ApoB100 on its surface[2][5], and are secreted into the circulation to be distributed among the other tissues (Figure 2).
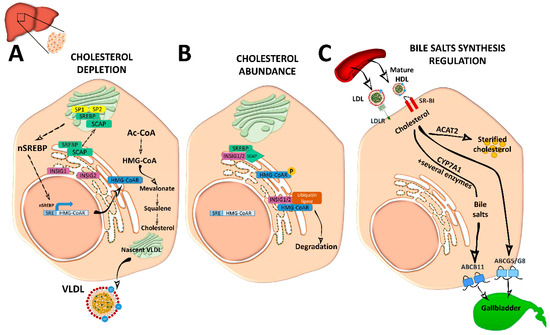
Figure 2. Cholesterol and bile acid biosynthesis. (A) In conditions of cholesterol depletion, the sterol regulatory element-binding protein 2 (SREBP2)–SREBP cleavage activating protein (SCAP) complex in the ER is transported to the Golgi apparatus where it is sequentially cleaved by site 1 protease (SP1) and SP2 at different sites to become active. Active nuclear (n)SREBP2 enters into the nucleus and binds to sterol regulatory element (SRE) sequences of target genes to induce the expression of genes involved in cholesterol synthesis such as the rate-limiting cholesterol synthesis enzyme 3-hydroxy-3-methyl-glutaryl coenzyme A reductase (HMG-CoAR). De novo synthetized cholesterol is stored or assembled in very low density lipoproteins (VLDL) in the Golgi apparatus for secretion into the circulation. (B) When ER membrane cholesterol exceeds 5% mol of the total lipids, sterol-sensing domains (SSD) in SCAP undergo a conformational change and the complex binds to insulin-induced gene 1 (INSIG1) or INSIG2, remaining inactive. Meanwhile, HMG-CoAR also binds to INSIG1 or INSIG2 for ubiquitilation and degradation, or it is inactivated by phosphorylation. (C) Cholesterol enters into the hepatocyte by low-density lipoprotein (LDL) receptor (LDLr) or through scavenger receptor class B type 1 (SRBI) by the delivery of high-density lipoprotein (HDL) reverse cholesterol transport. In the liver, it is stored as cholesterol ester after being esterified by acetyl coenzyme A (Ac-CoA) acetyltransferase 2 (ACAT2). It can also be used to synthetize bile acids in the biosynthetic metabolic pathway with the cytochrome P450 family 7 subfamily A member 1 (CYP7A1) enzyme as a rate-limiting factor. Bile acids will be secreted into the gallbladder as bile salts by adenosine triphosphate-binding cassette subfamily B member 11 (ABCB11) transporter. Cholesterol excess is also effluxed by adenosine triphosphate-binding cassette transporter G5/G8 (ABCG5/G8) into the gallbladder for secretion into the intestine.
3.3. Cholesterol Transport and Lipoprotein Metabolism
In the bloodstream, triglycerides in chylomicrons, very low density chylomicrons, and VLDL are hydrolyzed by lipoprotein lipase (LPL) enzyme, present in the endothelial surface of capillaries of most tissues and will be used as a source of energy or stored intracellularly[2]. Cholesteryl ester-rich chylomicron remnants are taken up by the liver and its components are incorporated in the newly synthetized VLDL particles[1]. VLDL will become first intermediate-density lipoproteins (IDL) and via hepatic lipase (HL) action will generate low-density lipoproteins (LDL). These lipoproteins can be further catabolized by lipases, a process that increases their density. In addition, VLDL and LDL can also be enriched in cholesterol by the cholesteryl ester transfer protein (CETP), which transfers cholesteryl esters from high-density lipoprotein (HDL)[2]. Resultant LDL are cleared by the LDL receptor (LDLr) in different tissues[9](Figure 1). LDL-bound LDLr undergoes the clathrin-mediated endocytosis and cholesteryl esters will enter into the endolysosomal system to transport cholesterol to the ER. LDLr returns to the plasma membrane by an endosomal recycling complex or undergoes lysosomal degradation through binding to proprotein convertase proprotein convertase subtilisin kexin 9 (PCSK9), which is strongly upregulated by SREBP and downregulated by insulin/mammalian target of rapamycin (mTOR)c signaling[3]. LDLr can also enter in lysosomal degradation by polyubiquitylation-inducible degrader of LDLr (IDOL) E3 ligase (also called MYLIP).
3.4. Cellular Cholesterol Efflux and Storage
Mature spherical lipid-rich HDL returns by the reverse cholesterol transport to the liver where it delivers cholesterol through the SRBI for excretion (Figure 1)[4]. HDLs are the densest and smallest particles with a high protein content of mostly ApoA-I protein. ABCA1 and lipid-free ApoA-I are responsible for removing cholesterol excess from cells and form nascent discoidal HDL particles that contain ApoA-I. Once released into the circulation, nascent HDL becomes spherical mature HDL by LCAT, CETP, and HL action[3][9](Figure 1). ApoA-I can also be endocyted in clathrin-coated vesicles and directly receive LDL-derived cholesterol from late endosomes via Niemann-Pick type c (NPC)2. Notably, in macrophages, ABCA1 and ABCG1 have a major role in cholesterol removal by preventing foam cell formation. ABCA1 and ABCG1 are upregulated by retinoid X receptor (RXR) and liver X receptor (LXR). Transactivation of ABCA1 is also upregulated by LXR-responsive long non-coding RNA (lncRNA) called MeXis[3]. Back in the liver, HDL-derived sterols bind to taurine, glycine, or sulfate, generating bile salts that are secreted by ABCB11 transporter to the canalicular membrane to form bile salt-phosphatidylcholine micelles[1][3](Figure 2). This HDL-mediated pathway is key in cholesterol metabolism as it is the main pathway of cholesterol removal from the body. Hepatic ABCG5 and ABCG8, a heterodimeric transporter (ABCG5/G8) positively regulated by LXR, excretes excess neutral sterol into the bile[1][3][10](Figures 1 and 2). After food ingestion, bile stored in the gallbladder is secreted into the intestine where it is reabsorbed by membrane protein NPC1L1 or excreted with the feces. In the liver, bile acids also act as ligands of several nuclear receptors such as FXR and pregnane X receptor (PXR)[1]. FXR also functions as a bile acid sensor, regulating bile acid synthesis genes that inhibit CYP7A1, a rate-limiting enzyme in bile acid synthesis[1][10][11](Figure 2).
Intracellular cholesterol is esterified by two membrane enzymes, ACAT1 and ACAT2[3]. ACAT1, found predominantly in macrophages, epithelial cells, and steroid hormone-producing cells, is activated by cholesterol and by two promoters, P1 and P7[3]. In enterocytes and hepatocytes, ACAT2 is potently activated by cholesterol and has a greater affinity for 25-hydroxycholesterol and bile acid derivatives. ACAT2 gene expression is regulated by several transcription factors, including hepatocyte nuclear factor 1α (HNF1α), HNF4α and caudal type homeobox 2 (CDX2), and by proteasomal degradation through ubiquitylation[3] (Figures 1 and 2).
References
- Jan Freark De Boer; Folkert Kuipers; Albert K. Groen; Cholesterol Transport Revisited: A New Turbo Mechanism to Drive Cholesterol Excretion. Trends in Endocrinology & Metabolism 2018, 29, 123-133, 10.1016/j.tem.2017.11.006.
- Chih-Wei Ko; Jie Qu; Dennis D. Black; Patrick Tso; Regulation of intestinal lipid metabolism: current concepts and relevance to disease. Nature Reviews Gastroenterology & Hepatology 2020, 17, 169-183, 10.1038/s41575-019-0250-7.
- Jie Luo; Hongyuan Yang; Bao-Liang Song; Mechanisms and regulation of cholesterol homeostasis. Nature Reviews Molecular Cell Biology 2019, 21, 225-245, 10.1038/s41580-019-0190-7.
- Milessa Silva Afonso; Roberta Marcondes Machado; Ana Maria Lottenberg; Eder C.R. Quintão; Kathryn J. Moore; Ana Maria Lottenberg; Molecular Pathways Underlying Cholesterol Homeostasis. Nutrients 2018, 10, 760, 10.3390/nu10060760.
- Elina Ikonen; Cellular cholesterol trafficking and compartmentalization. Nature Reviews Molecular Cell Biology 2008, 9, 125-138, 10.1038/nrm2336.
- Joseph L Goldstein; Michael S. Brown; A century of cholesterol and coronaries: from plaques to genes to statins.. Cell 2015, 161, 161-172, 10.1016/j.cell.2015.01.036.
- Arun Radhakrishnan; Joseph L. Goldstein; Jeffrey G. McDonald; Michael S. Brown; Switch-like Control of SREBP-2 Transport Triggered by Small Changes in ER Cholesterol: A Delicate Balance. Cell Metabolism 2008, 8, 512-521, 10.1016/j.cmet.2008.10.008.
- Laura J. Sharpe; Emma Cook; Noam Zelcer; Andrew J. Brown; The UPS and downs of cholesterol homeostasis. Trends in Biochemical Sciences 2014, 39, 527-535, 10.1016/j.tibs.2014.08.008.
- Leigh Goedeke; Carlos Fernández-Hernando; Regulation of cholesterol homeostasis. Cellular and Molecular Life Sciences 2011, 69, 915-930, 10.1007/s00018-011-0857-5.
- Anna C. Calkin; Peter Tontonoz; Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nature Reviews Molecular Cell Biology 2012, 13, 213-224, 10.1038/nrm3312.
- Claire Mazuy; Audrey Helleboid; Bart Staels; Philippe Lefebvre; Nuclear bile acid signaling through the farnesoid X receptor. Cellular and Molecular Life Sciences 2014, 72, 1631-1650, 10.1007/s00018-014-1805-y.

