 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Swee T Tan | + 3063 word(s) | 3063 | 2021-08-20 11:20:12 | | | |
| 2 | Amina Yu | -1 word(s) | 3062 | 2021-09-08 11:14:22 | | |
Video Upload Options
The Renin-angiotensin system (RAS), as a constituent of the tumor microenvironment (TME), is involved in several hallmarks of cancer, including angiogenesis, hypoxia, and tumor cell proliferation. Components of the RAS are expressed in different types of cancer including colon adenocarcinoma and malignant melanoma. RAS components are also expressed by cancer stem cells in oral cavity squamous cell carcinoma (SCC), renal clear cell carcinoma, primary, and metastatic, cutaneous SCC, metastatic colon adenocarcinoma, metastatic malignant melanoma, and glioblastoma.
1. Introduction
Glioblastoma (GB), the most common and most aggressive primary brain cancer in humans, is classified as a WHO grade IV astrocytoma, and is characterized by microvascular proliferation and central necrosis[1]. Primary GB arises de novo and accounts for 90% of cases with a predilection for older individuals, while secondary GB arises from low-grade astrocytoma and affects younger patients[2] . GB has been categorized into four distinct molecular subtypes: classical, mesenchymal, neural, and proneural[3] , although other studies have only identified classical, mesenchymal, and proneural subtypes [4]. The classical subtype includes amplification or mutation of epidermal growth factor receptor, the mesenchymal subtype includes deletions of the 17q11.2 region containing the gene NF1, and the proneural subtype is characterized by high levels of platelet-derived growth factor receptor α expression and point mutations in isocitrate dehydrogenase 1 (IDH1) and p53[3].
Various genetic or epigenetic changes may affect the prognosis of GB patients including IDH mutations and O6-methylguanine-DNA methyltransferase (MGMT) methylation status. GB may be divided into IDH-wild-type and IDH-mutant tumors. IDH is an enzyme that catalyzes oxidative decarboxylation of isocitrate to 2-oxoglutarate. The most common mutation in GB affects IDH1 with a single amino acid missense mutation at arginine 132 which is replaced by histidine[5]. IDH-wild-type GB is more common, tends to arise de novo, and is generally more aggressive with a worse prognosis than IDH-mutant GB. By contrast, IDH-mutant GB is predominantly observed in secondary GB and is associated with a better prognosis[6] . The current standard treatment for GB involves maximal safe surgical resection with adjuvant chemotherapy and radiotherapy, known as the Stupp protocol [7].Temozolomide, an alkylating agent, is used as first-line chemotherapy for GB with its efficacy related to the methylation status of the MGMT promoter[8] =. MGMT methylation is associated with an improved overall survival in GB patients [9]. Despite this intensive treatment, tumor recurrence in GB patients is inevitable with an overall median survival time of 14.6 months with a range of 12–14 months which has not changed since the introduction of the Stupp protocol in 2005 [10][11].
2. GB Tumor Microenvironment
The GB tumor microenvironment (TME) is highly heterogeneous and consists of cancer cells and non-cancer cells. Non-cancer cell types include immune cells, such as tumor-associated macrophages (TAMs), resident glial cells, peripheral macrophages, endothelial cells, pericytes, astrocytes, cancer stem cells (CSCs), fibroblasts, and other components such as the extracellular matrix [12]. Given the rarity of extracranial metastasis from GB [13], it appears that GB development requires the unique intracerebral microenvironment inclusive of the blood–brain barrier (BBB) [14]. The TME, with emphasis on glioma-associated microglia/macrophages, pericytes, and reactive astrocytes, is increasingly recognized to play a critical role in GB development and progression [15]. The idea that cytokines, growth factors, chemokines, inflammatory mediators, and remodeling enzymes are involved in intra- and inter-cellular communications within the TME is not novel [16]. Additionally, constant communication between GB cells and the surrounding TME [14] is facilitated by extracellular vesicles that expedite bi-directional cross-talk within the TME [12][17].
Anatomically distinct regions of the TME, known as tumor niches, are thought to contain CSCs and play a fundamental role in the regulation of metabolism, immune surveillance, survival, invasion, and self-maintenance with the renin–angiotensin system (RAS) playing a critical role [12][18][19]. The GB TME may consist of several distinct tumor niches including the hypoxic tumor niche, the perivascular or angiogenic tumor niche, and the vascular-invasive tumor niche. The perivascular niche contains CSCs in close juxtaposition with the abnormal angiogenic vasculature and provides a supportive environment for CSC growth, maintenance, and survival. The vasculature in the hypoxic tumor niche is either non-functional or has regressed, leading to areas of necrosis that are surrounded by rows of hypoxic palisading tumor cells [20]. The vascular-invasive tumor niche contains tumor cells co-opted with normal blood vessels that migrate deep into the brain parenchyma [20]
GB is highly vascular and is characterized by extensive neovascularization and pathological angiogenesis predominantly induced by vascular endothelial growth factor (VEGF), which is produced by tumor cells, CSCs, and immune cells [21][22] Other angiogenic factors, such as transforming growth factor-β1, platelet-derived growth factor-BB, and fibroblast growth factor-2, may also play a role in the pathological angiogenesis [23][24]. In addition to endothelial proliferation, bone marrow-derived endothelial and pericyte progenitor cells may be recruited and incorporated into the growing vessels[25]. There is also evidence that CSCs may be involved in neovascularization by differentiating into endothelial cells or pericytes in GB [26][27][28]. Increased VEGF expression also fosters an immunosuppressive microenvironment that enables tumors, including GB, to evade host immune surveillance [29]. The abnormal vasculature in GB includes dilated and leaky vessels and glomeruloid microvascular proliferation in which endothelial cells and pericytes form poorly organized vascular structures, which effectively disrupt the BBB, leading to cerebral edema. In addition, the blood–brain tumor barrier hinders drug delivery to the tumor [30].
The BBB is a highly specialized, selectively permeable barrier between the brain and the systemic blood supply that helps to maintain homeostasis of the cerebral microenvironment. The structure of the BBB includes endothelial cells with tight junctions, adherens junctions, astrocytes, pericytes, and the basement membrane [31]. The BBB plays several fundamental roles, including supplying the brain with essential nutrients, such as oxygen and glucose, mediating the efflux of waste products, facilitating the movement of nutrients and plasma proteins, and restricting toxins into the central nervous system[32]. Disruption of the BBB and its tight regulation of the cerebral microenvironment leads to increased blood vessel permeability with plasma and fluid leakage into the tumor tissue causing cerebral edema and raised interstitial and intracranial pressure[33]. The combination of abnormal vasculature in GB and the disruption of the BBB leads to impaired blood flow and reduced oxygen delivery within the tumor [34]. Microvascular thrombosis may also occur causing occlusion of the blood vessels, further promoting intra-tumoral hypoxia, leading to pseudo-palisading necrosis[35]. Hypoxia is also a consequence of increased oxygen diffusion distance due to the fact of tumor growth and expansion[34], which may, in and of itself, be a key regulator of tumor cell survival, stemness, and immune surveillance in the TME [36][37][38]. Hypoxia also sustains tumor cell proliferation, invasiveness, and contributes to chemotherapy and radiotherapy resistance. This occurs via inhibition of free radicals, which reduces the efficacy of radiotherapy[39], and through upregulation of the multi-drug resistance gene, MDR1/ABCB1 , which reduces chemotherapy effectiveness. Hypoxia-inducible factor-1 (HIF-1) and HIF-2 mediate the response to hypoxia on a molecular level in GB [40] and may potentially modify CSCs [41]. The GB microenvironmental niche also consists of pseudo-palisading glioma cells that upregulate HIF proteins, inducing expression of factors, such as VEGF and interleukin 8, which are implicated in tumor cell survival, metabolism, invasion, and angiogenesis. The resultant cross-talk releases pro-inflammatory signals from the areas of necrosis in the hypoxic tumor niche into the surrounding TME, promoting immunosuppression, and angiogenesis [42].
Immune cells, including circulating monocytes, neutrophils, and myeloid-derived suppressor cells (MDSCs), are another source of angiogenic factors. In ovarian cancer, MDSCs increase CSC characteristics by increasing microRNA-101 expression, which induces the expression of stemness genes [43]. It is interesting to speculate that MDSCs also regulate the stemness of CSCs within the GB TME via this mechanism. These cells may enter the brain as a result of breakdown of the BBB in GB and the production of tumor-derived chemokines and cytokines, contributing to the immunosuppressive GB TME [44–46]. TAMs are the dominant immune cell population in GB and may include resident microglial cells and peripheral macrophages [47,48]. Traditionally, TAMs have been defined as either anti-tumoral M1/Th1 (classical-activated macrophages) or protumoral M2/Th2 (alternative-activated macrophages) phenotypes. M1 macrophages foster the inflammatory response by secreting pro-inflammatory cytokines such as IL-12, tumor necrosis factor-α (TNF-α), CXCL-10, and interferon-γ (IFN-γ) and produce high levels of nitric oxide synthase to exert anti-tumor cell activity (ref)(Figure 1). M2 macrophages, on the other hand, play a key immunosuppressive function by secreting anti-inflammatory cytokines, such as IL-10, IL-13, and IL-4, and express abundant arginase-1, mannose receptor CD206, and scavenger receptors to promote tumor progression [49–51]. The release of TGF-β by TAMs has been shown to induce matrix metalloproteinase 9 (MMP9) and, thus, increase CSC invasiveness [52]. A more recent study has demonstrated that the TAM population is in a constant state of transition or plasticity between the two phenotypes and that M1 phenotype expression may be enhanced by TME changes or therapeutic interventions [51]. Resident microglia are present within the brain, but it is the recruitment of peripheral macrophages to the GB TAM pool, in particular, that may mediate tumor phagocytosis with disruption of the signal regulatory protein α receptor (SIRP-α)– CD47 axis. This facilitates immune evasion because the antiphagocytic “don’t eat me” surface protein CD47 is upregulated, which binds to SIRP-α on phagocytic cells to inhibit phagocytosis [53]. However, even in the absence of macrophages, resident microglia may be transformed into effector cells of tumor cell phagocytosis, in response to anti-CD47 blockade [54]. In models of pancreatic ductal adenocarcinoma, for example, RP-182 may selectively induce conformational switching of the mannose receptor CD206, which is expressed on the M2 TAM phenotype, ultimately reprogramming M2-like TAMs into an anti-tumor M1-like phenotype [55]. The immunosuppressive phenotype of TAMs may be controlled by long-chain fatty acid metabolism, and chemical inhibitors targeting this metabolic pathway may block TAM polarization in vitro and tumor growth in vivo [56]. GB-derived exosomes may reprogram M1 macrophages to M2 macrophages and condition M2 macrophages to become strongly immunosuppressive TAMs [57].
3. Glioblastoma Cancer Stem Cells
The CSC concept proposes that a small distinct population of cells within a tumor with self-renewal capability are responsible for driving tumorigenesis [43][44]. These CSCs may be defined as stem cell-like cells within a tumor that also have the capacity for proliferation and multi-potency. This may be regarded as a functional definition insofar as CSCs may be characterized through the generation of serially transplantable tumors that faithfully recapitulate the parent tumor [45]. There is marked intra- and inter-tumoral heterogeneity including, differing numbers of highly tumorigenic CSCs [46]. Such heterogeneity may be best explained by a combination of different models of cancer, including the stochastic model (also known as the clonal evolution model), the CSC concept of cancer (also known as the hierarchical model of cancer), and the concept of plasticity [47][48].
The traditional model of cancer is predicated on the stochastic model of carcinogenesis which proposes that cancer cells are derived from normal cells that acquire genetic and/or epigenetic mutations. This typically results in unidirectional transitions from benign to malignant cells. These malignant tumor cells have unrestricted division capacities and their high mutation rates increase the likelihood of successive generations of cloned cells being adapted to the selection pressures of the tumor site. However, the stochastic model does not fully account for all aspects of cancer biology including tumor recurrence following treatment [49].
In contrast, the CSC concept of cancer proposes that CSCs contribute to carcinogenesis, invasion, metastasis, therapy resistance, and recurrence [50][51]. CSCs divide asymmetrically into non-tumorigenic cancer cells, which form the bulk of a tumor, and identical highly tumorigenic but less abundant CSCs, which sit at the apex of the cellular hierarchy [52]. CSCs have been postulated to originate from non-malignant stem cells or progenitor cells [51] or dedifferentiated cancer cells [53]. The overlap between the stochastic model and the CSC concept may be explained by the concept of cellular plasticity whereby cancer cells may reversibly transition between stem-like and non-stem-like cell states [54]. This process of transition may be driven by embryonic stem cell (ESC)-associated regulatory networks and may be affected by the dynamic TME including the CSC niche [55]. Moreover, certain cancer cells may de-differentiate and re-enter the CSC pool, thus regaining the capacity for tumorigenesis and clonal expansion[56].
A crucial function of stem cells is self-renewal, for which the Notch, Sonic hedgehog, and Wnt signaling pathways may be essential[57] . GB expresses a number of stemness-associated markers including cell surface markers (CD133, CD15, A2B5, and L1CAM), cytoskeletal proteins (nestin), transcription factors (SOX2, NANOG, and OCT4), post-transcriptional factors, and polycomb transcriptional suppressors (Bmi1 and Ezh2) [58]. There is also evidence of plasticity and bi-directional interconversion between CSCs and cancer cells [59]. In a landmark study, pluripotent stem cells were formed from reprogrammed mouse embryonic and adult fibroblasts by the addition of transcription factors OCT4, SOX2, c-MYC, and KLF4[60] . These factors, in addition to NANOG, which are expressed by ESCs, have been identified in GB[61] . The capacity of GB cells for perpetual self-renewal may rely on the contribution from transcription factors such as OCT4 and SOX2[58] . SOX2 is highly expressed in GB[61] and may play a key role in maintaining plasticity for bi-directional cellular conversion in GB[62]. Moreover, silencing of SOX2 inhibits tumor proliferation in GB [63] and, thus, it may be a potential therapeutic target in the treatment of GB[64] . Another potential therapeutic target involves the JAK–STAT3 signaling pathway which is also associated with the self-renewal capacity of GB. Inhibition of this pathway may impede the migratory and invasive potential of GB by decreasing activation of the transcription factor STAT3 and, thus, reducing the levels of matrix metalloproteinases and associated invadopodia activity[65]. In addition, STAT3 binding to the Notch1 promoter inhibits this signaling pathway and may impede the maintenance of glioma stem-like cells while reducing the expression of glioma stem cell markers CD133, SOX2, and nestin[66] (Figure1).
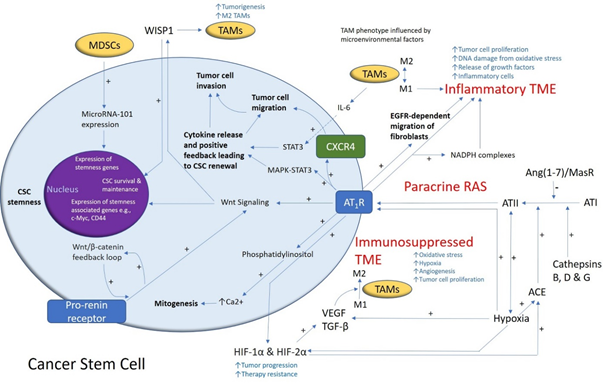
Figure1. A schema demonstrating the role of the renin–angiotensin system (RAS) and its convergent signaling pathways in the glioblastoma tumor microenvironment (TME) and cancer stem cells (CSCs). A cancer stem cell (with the cytoplasm depicted in light blue and the nucleus in purple) residing within the glioblastoma TME. Angiotensin II (ATII), the physiologically active end-product of the paracrine RAS, activates ATII receptor 1 (AT1R) leading to increased tumor cell proliferation, oxidative stress, hypoxia and angiogenesis, and inflammation—the hallmarks of cancer. This contributes to an inflammatory TME by increasing the number of inflammatory cells, partly by increasing the number of NADPH complexes, leading to tumor cell proliferation, DNA damage from oxidative stress, and release of growth factors. AT1R also activates phosphatidylinositol signaling, which increases cytosolic Ca2+ to promote mitogenesis. Hypoxia increases paracrine RAS activity by upregulating angiotensin-converting enzyme (ACE) and the expression of hypoxia-inducible factor 1α (HIF-1α) and HIF-2α, which increase tumor progression and treatment resistance. HIF-1α, HIF-2α, and hypoxia increase the expression of vascular endothelial growth factor (VEGF) which increases angiogenesis. AT1R, via MAPK-STAT3 signaling, contributes to a cytokine release that leads to CSC renewal. C-X-C chemokine receptor type 4 (CXCR4) promotes tumor cell migration and invasion. AT1R signaling and the prorenin receptor, which act in a feedback loop with Wnt/β-catenin, increase Wnt signaling which promotes CSC stemness by upregulating stemness-associated markers. Myeloid-derived suppressor cells (MDSCs) promote CSC characteristics by increasing microRNA-101 expression that induces expression of stemness-related genes in CSCs. The Ang(1–7)/MasR axis opposes the ACE/ATII/AT1R axis. Cathepsins B, D, and G act as bypass loops for the RAS. Under the influence of the TME, polarization of tumor-associated macrophages (TAMs)—immune cells that are located within the TME—changes from the M1 to M2 phenotype. M2 TAMs induce the proliferation of CSCs via interleukin 6 (IL-6)-induced activation of STAT3, leading to cytokine release and positive feedback contributing to CSC renewal. Glioblastoma CSCs secrete Wnt-induced signaling protein 1 (WISP1), which facilitates a pro-tumor TME by promoting the survival of CSCs and M2 TAMs, and also promotes CSC maintenance. Abbreviations: ATI, angiotensin I; AT2R, ATII receptor 2; Ang(1–7), angiotensin 1–7; ATIII, angiotensin III; MAPK, mitogen-activated protein kinase. Figure modified and reproduced with permission from J Histochem Cytochem [19].
4. The Renin–Angiotensin System and Convergent Signaling Pathways in Glioblastoma
The RAS has been proposed to play an important role in the TME[19] in various cancer types, including lung cancer, through its effect on tumor cells, non-malignant cells, hypoxia, angiogenesis, and the inflammatory response [67]. The RAS is a complex physiological system and has a multitude of interactions with many different convergent signaling pathways that operate in carcinogenesis.
The RAS, as a constituent of the TME, is involved in several hallmarks of cancer, including angiogenesis, hypoxia, and tumor cell proliferation [68]. Components of the RAS are expressed in different types of cancer including colon adenocarcinoma[69] and malignant melanoma[70]. RAS components are also expressed by CSCs in oral cavity SCC [71][72], renal clear cell carcinoma[73], primary[74] , and metastatic[75], cutaneous SCC, metastatic colon adenocarcinoma[76] , metastatic malignant melanoma [77][78], and GB[79]. In GB, prorenin receptor (PRR), AT1R, and angiotensin II receptor 2 are co-expressed with stemness-associated markers [79]. PRR is highly expressed in GB compared with lower-grade gliomas; this higher expression of PRR in higher-grade glioma is notable as the Wnt/β-catenin signaling pathway is implicated in the self-renewal of stem cells [80].
The Wnt/β-catenin signaling pathway, which sits downstream of the RAS, is implicated in tumor initiation in several cancer types [81] In brief, this pathway results in active β-catenin translocating into the nucleus, upregulating the expression of oncogenes such as c-MYC, AXIN2, and CCND1 [81]. PRR is a component of the Wnt receptor complex and acts as an adapter between vacuolar H + -adenosine triphosphate (V-ATPase) and low-density lipoprotein receptor-related protein 6. V-ATPase, a proton pump, is essential for cellular acidification and is involved in the mechanism for β-catenin activation [82]. This process facilitates binding of Wnts to their respective Wnt receptor complex [83]. Further, PRR promotes brain cancers via the Wnt/β-catenin signaling pathway, and in addition to being a membrane receptor, exists in the cytoplasm and increases the protein expression of Wnt2 within glioma cells[80]. This evidence underscores the PRR as a potential oncoprotein via Wnt/β-catenin pathway-related carcinogenesis[81], which influences cell stemness [84], tumorigenesis, and cellular proliferation [85][86]. Renin is expressed in GB and may contribute to the mechanisms of neovascularization in GB[87]. Furthermore, downregulation of the Ang(1–7)/MAS signaling axis by podocalyxin results in enhanced GB cell invasion and proliferation [88]. Finally, bypass loops of the RAS involving various cathepsins that may also contribute to the proliferative activity in GB, for example, cathepsin G coverts ATI to AII and from AGT directly to ATII, which binds to AT 1R, to promote cancer progression[89][90][91]. GB CSCs have been shown to secrete Wnt-induced signaling protein 1 that promotes the survival of both the CSCs and M2 TAMs to promote a pro-TME[92] .
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Crespo, I.; Vital, A.L.; Gonzalez-Tablas, M.; Patino Mdel, C.; Otero, A.; Lopes, M.C.; De Oliveira, C.; Domingues, P.; Orfao, A.; Tabernero, M.D. Molecular and Genomic Alterations in Glioblastoma Multiforme. Am. J. Pathol. 2015, 185, 1820–1833. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; DeCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e46. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Zentgraf, H.; Balss, J.; Hartmann, C.; Von Deimling, A. Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol. 2009, 118, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Garrett, M.; Fujii, Y.; Osaka, N.; Ito, D.; Hirota, Y.; Sasaki, A.T. Emerging Roles of Wild-type and Mutant IDH1 in Growth, Metabolism and Therapeutics of Glioma. In Gliomas; Debinski, W., Ed.; Exon Publications: Brisbane, Australia, 2021. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Binabaj, M.M.; Bahrami, A.; ShahidSales, S.; Joodi, M.; Joudi Mashhad, M.; Hassanian, S.M.; Anvari, K.; Avan, A. The prognostic value of MGMT promoter methylation in glioblastoma: A meta-analysis of clinical trials. J. Cell Physiol. 2018, 233, 378–386. [Google Scholar] [CrossRef]
- Das, S.; Marsden, P.A. Angiogenesis in glioblastoma. N. Engl. J. Med. 2013, 369, 1561–1563. [Google Scholar] [CrossRef]
- Gately, L.; McLachlan, S.A.; Dowling, A.; Philip, J. Life beyond a diagnosis of glioblastoma: A systematic review of the literature. J. Cancer Surviv. 2017, 11, 447–452. [Google Scholar] [CrossRef]
- Simon, T.; Jackson, E.; Giamas, G. Breaking through the glioblastoma micro-environment via extracellular vesicles. Oncogene 2020, 39, 4477–4490. [Google Scholar] [CrossRef] [PubMed]
- Awan, M.; Liu, S.; Sahgal, A.; Das, S.; Chao, S.T.; Chang, E.L.; Knisely, J.P.; Redmond, K.; Sohn, J.W.; Machtay, M.; et al. Extra-CNS metastasis from glioblastoma: A rare clinical entity. Expert Rev. Anticancer Ther. 2015, 15, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Da Ros, M.; De Gregorio, V.; Iorio, A.L.; Giunti, L.; Guidi, M.; De Martino, M.; Genitori, L.; Sardi, I. Glioblastoma Chemoresistance: The Double Play by Microenvironment and Blood-Brain Barrier. Int. J. Mol. Sci. 2018, 19, 2879. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and Niche Concept. Cancers 2018, 11, 5. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef]
- Sullivan, R.; Maresh, G.; Zhang, X.; Salomon, C.; Hooper, J.; Margolin, D.; Li, L. The Emerging Roles of Extracellular Vesicles As Communication Vehicles within the Tumor Microenvironment and Beyond. Front. Endocrinol. 2017, 8, 194. [Google Scholar] [CrossRef]
- Haznedaroglu, I.C.; Malkan, U.Y. Local bone marrow renin-angiotensin system in the genesis of leukemia and other malignancies. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4089–4111. [Google Scholar]
- Kilmister, E.J.; Tan, S.T. The Role of the Renin-Angiotensin System in the Cancer Stem Cell Niche. J. Histochem. Cytochem. 2021, 00221554211026295. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiang, W.; Xue, B.Z.; Yi, D.Y.; Zhao, H.Y.; Fu, P. Growth factors contribute to the mediation of angiogenic capacity of glioma-associated mesenchymal stem cells. Oncol. Lett. 2021, 21, 215. [Google Scholar] [CrossRef] [PubMed]
- Monteforte, A.; Lam, B.; Sherman, M.B.; Henderson, K.; Sligar, A.D.; Spencer, A.; Tang, B.; Dunn, A.K.; Baker, A.B. (*) Glioblastoma Exosomes for Therapeutic Angiogenesis in Peripheral Ischemia. Tissue Eng. Part A 2017, 23, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegue, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef]
- Tamura, R.; Tanaka, T.; Akasaki, Y.; Murayama, Y.; Yoshida, K.; Sasaki, H. The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: Perspectives for therapeutic implications. Med. Oncol. 2019, 37, 2. [Google Scholar] [CrossRef] [PubMed]
- Van Tellingen, O.; Yetkin-Arik, B.; De Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; De Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro. Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef]
- Rong, Y.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. ‘Pseudopalisading’ necrosis in glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 529–539. [Google Scholar] [CrossRef]
- Seidel, S.; Garvalov, B.K.; Wirta, V.; Von Stechow, L.; Schanzer, A.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.T.; Nister, M.; et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 2010, 133, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef]
- Bar, E.E.; Lin, A.; Mahairaki, V.; Matsui, W.; Eberhart, C.G. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am. J. Pathol. 2010, 177, 1491–1502. [Google Scholar] [CrossRef]
- Guardia, G.D.A.; Correa, B.R.; Araujo, P.R.; Qiao, M.; Burns, S.; Penalva, L.O.F.; Galante, P.A.F. Proneural and mesenchymal glioma stem cells display major differences in splicing and lncRNA profiles. NPJ Genom. Med. 2020, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Zagzag, D.; Lukyanov, Y.; Lan, L.; Ali, M.A.; Esencay, M.; Mendez, O.; Yee, H.; Voura, E.B.; Newcomb, E.W. Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: Implications for angiogenesis and glioma cell invasion. Lab. Investig. 2006, 86, 1221–1232. [Google Scholar] [CrossRef]
- Bar, E.E. Glioblastoma, cancer stem cells and hypoxia. Brain Pathol. 2011, 21, 119–129. [Google Scholar] [CrossRef]
- Filatova, A.; Acker, T.; Garvalov, B.K. The cancer stem cell niche(s): The crosstalk between glioma stem cells and their microenvironment. Biochim. Biophys. Acta 2013, 1830, 2496–2508. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef]
- Adams, J.M.; Strasser, A. Is tumor growth sustained by rare cancer stem cells or dominant clones? Cancer Res. 2008, 68, 4018–4021. [Google Scholar] [CrossRef]
- Rich, J.N. Cancer stem cells: Understanding tumor hierarchy and heterogeneity. Medecine 2016, 95, S2–S7. [Google Scholar] [CrossRef]
- Najafi, M.; Mortezaee, K.; Ahadi, R. Cancer stem cell (a)symmetry & plasticity: Tumorigenesis and therapy relevance. Life Sci. 2019, 231, 116520. [Google Scholar] [CrossRef]
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef]
- Gurel, C.; Inetas, G.; Hortu, I.; Tunc, E.; Kuscu, G.C.; Dindaroglu, F.C.; Sahin, O.; Buhur, A.; Oktem, G. Cancer and Cancer Stem Cells: New Molecular Perspectives. Crit. Rev. Oncog. 2019, 24, 99–104. [Google Scholar] [CrossRef]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef]
- Bradshaw, A.; Wickremsekera, A.; Tan, S.T.; Peng, L.; Davis, P.F.; Itinteang, T. Cancer Stem Cell Hierarchy in Glioblastoma Multiforme. Front. Surg. 2016, 3, 21. [Google Scholar] [CrossRef]
- Eun, K.; Ham, S.W.; Kim, H. Cancer stem cell heterogeneity: Origin and new perspectives on CSC targeting. BMB Rep. 2017, 50, 117–125. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Quail, D.F.; Taylor, M.J.; Postovit, L.M. Microenvironmental regulation of cancer stem cell phenotypes. Curr. Stem Cell Res. Ther. 2012, 7, 197–216. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. How does multistep tumorigenesis really proceed? Cancer Discov. 2015, 5, 22–24. [Google Scholar] [CrossRef]
- Iwadate, Y. Plasticity in Glioma Stem Cell Phenotype and Its Therapeutic Implication. Neurol. Med. Chir. 2018, 58, 61–70. [Google Scholar] [CrossRef]
- Kalkan, R. Glioblastoma Stem Cells as a New Therapeutic Target for Glioblastoma. Clin. Med. Insights. Oncol. 2015, 9, 95–103. [Google Scholar] [CrossRef]
- Safa, A.R.; Saadatzadeh, M.R.; Cohen-Gadol, A.A.; Pollok, K.E.; Bijangi-Vishehsaraei, K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. 2015, 2, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, A.; Wickremesekera, A.; Brasch, H.D.; Chibnall, A.M.; Davis, P.F.; Tan, S.T.; Itinteang, T. Cancer Stem Cells in Glioblastoma Multiforme. Front. Surg. 2016, 3, 48. [Google Scholar] [CrossRef]
- Berezovsky, A.D.; Poisson, L.M.; Cherba, D.; Webb, C.P.; Transou, A.D.; Lemke, N.W.; Hong, X.; Hasselbach, L.A.; Irtenkauf, S.M.; Mikkelsen, T.; et al. Sox2 promotes malignancy in glioblastoma by regulating plasticity and astrocytic differentiation. Neoplasia 2014, 16, 193–206.e25. [Google Scholar] [CrossRef]
- Gangemi, R.M.; Griffero, F.; Marubbi, D.; Perera, M.; Capra, M.C.; Malatesta, P.; Ravetti, G.L.; Zona, G.L.; Daga, A.; Corte, G. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells 2009, 27, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Garros-Regulez, L.; Garcia, I.; Carrasco-Garcia, E.; Lantero, A.; Aldaz, P.; Moreno-Cugnon, L.; Arrizabalaga, O.; Undabeitia, J.; Torres-Bayona, S.; Villanua, J.; et al. Targeting SOX2 as a Therapeutic Strategy in Glioblastoma. Front. Oncol. 2016, 6, 222. [Google Scholar] [CrossRef] [PubMed]
- Senft, C.; Priester, M.; Polacin, M.; Schroder, K.; Seifert, V.; Kogel, D.; Weissenberger, J. Inhibition of the JAK-2/STAT3 signaling pathway impedes the migratory and invasive potential of human glioblastoma cells. J. Neurooncol. 2011, 101, 393–403. [Google Scholar] [CrossRef]
- Yahyanejad, S.; King, H.; Iglesias, V.S.; Granton, P.V.; Barbeau, L.M.; Van Hoof, S.J.; Groot, A.J.; Habets, R.; Prickaerts, J.; Chalmers, A.J.; et al. NOTCH blockade combined with radiation therapy and temozolomide prolongs survival of orthotopic glioblastoma. Oncotarget 2016, 7, 41251–41264. [Google Scholar] [CrossRef]
- Catarata, M.J.; Ribeiro, R.; Oliveira, M.J.; Robalo Cordeiro, C.; Medeiros, R. Renin-Angiotensin System in Lung Tumor and Microenvironment Interactions. Cancers 2020, 12, 1457. [Google Scholar] [CrossRef]
- Wegman-Ostrosky, T.; Soto-Reyes, E.; Vidal-Millan, S.; Sanchez-Corona, J. The renin-angiotensin system meets the hallmarks of cancer. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 227–233. [Google Scholar] [CrossRef]
- Beitia, M.; Solano-Iturri, J.D.; Errarte, P.; Sanz, B.; Perez, I.; Etxezarraga, M.C.; Loizate, A.; Asumendi, A.; Larrinaga, G. Altered expression of renin-angiotensin system receptors throughout colorectal adenoma-adenocarcinoma sequence. Int. J. Med. Sci. 2019, 16, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Renziehausen, A.; Wang, H.; Rao, B.; Weir, L.; Nigro, C.L.; Lattanzio, L.; Merlano, M.; Vega-Rioja, A.; Del Carmen Fernandez-Carranco, M.; Hajji, N.; et al. The renin angiotensin system (RAS) mediates bifunctional growth regulation in melanoma and is a novel target for therapeutic intervention. Oncogene 2019, 38, 2320–2336. [Google Scholar] [CrossRef]
- Featherston, T.; Yu, H.H.; Dunne, J.C.; Chibnall, A.M.; Brasch, H.D.; Davis, P.F.; Tan, S.T.; Itinteang, T. Cancer Stem Cells in Moderately Differentiated Buccal Mucosal Squamous Cell Carcinoma Express Components of the Renin-Angiotensin System. Front. Surg. 2016, 3, 52. [Google Scholar] [CrossRef] [PubMed]
- Itinteang, T.; Dunne, J.C.; Chibnall, A.M.; Brasch, H.D.; Davis, P.F.; Tan, S.T. Cancer stem cells in moderately differentiated oral tongue squamous cell carcinoma express components of the renin-angiotensin system. J. Clin. Pathol. 2016, 69, 942–945. [Google Scholar] [CrossRef]
- Siljee, S.; Milne, B.; Brasch, H.D.; Bockett, N.; Patel, J.; Davis, P.F.; Kennedy-Smith, A.; Itinteang, T.; Tan, S.T. Expression of Components of the Renin-Angiotensin System by Cancer Stem Cells in Renal Clear Cell Carcinoma. Biomolecules 2021, 11, 537. [Google Scholar] [CrossRef]
- Nallaiah, S.; Lee, V.M.Y.; Brasch, H.D.; De Jongh, J.; Schaijik, B.V.; Marsh, R.; Tan, S.T.; Itinteang, T. Cancer stem cells within moderately differentiated head and neck cutaneous squamous cell carcinoma express components of the renin-angiotensin system. J. Plast. Reconstr. Aesthet. Surg. 2019, 72, 1484–1493. [Google Scholar] [CrossRef]
- Siljee, S.; Buchanan, O.; Brasch, H.D.; Bockett, N.; Patel, J.; Paterson, E.; Purdie, G.L.; Davis, P.F.; Itinteang, T.; Tan, S.T. Cancer Stem Cells in Metastatic Head and Neck Cutaneous Squamous Cell Carcinoma Express Components of the Renin-Angiotensin System. Cells 2021, 10, 243. [Google Scholar] [CrossRef]
- Narayanan, A.; Wickremesekera, S.K.; Van Schaijik, B.; Marsh, R.W.; Brasch, H.D.; Tan, S.T.; Itinteang, T. Cancer stem cells in liver metastasis from colon adenocarcinoma express components of the renin-angiotensin system. J. Cancer Metastasis Treat. 2019, 5, 36. [Google Scholar] [CrossRef]
- Wickremesekera, A.C.; Brasch, H.D.; Lee, V.M.; Davis, P.F.; Parker, A.; Koeck, H.; Itinteang, T.; Tan, S.T. Cancer stem cell subpopulations in metastatic melanoma to the brain express components of the renin-angiotensin system. J. Cancer Metastasis Treat. 2019, 5, 62. [Google Scholar] [CrossRef]
- Yoganandarajah, V.; Patel, J.; Van Schaijik, B.; Bockett, N.; Brasch, H.D.; Paterson, E.; Sim, D.; Davis, P.F.; Roth, I.M.; Itinteang, T.; et al. Identification of Cancer Stem Cell Subpopulations in Head and Neck Metastatic Malignant Melanoma. Cells 2020, 9, 324. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, A.R.; Wickremesekera, A.C.; Brasch, H.D.; Chibnall, A.M.; Davis, P.F.; Tan, S.T.; Itinteang, T. Glioblastoma Multiforme Cancer Stem Cells Express Components of the Renin-Angiotensin System. Front. Surg. 2016, 3, 51. [Google Scholar] [CrossRef]
- Kouchi, M.; Shibayama, Y.; Ogawa, D.; Miyake, K.; Nishiyama, A.; Tamiya, T. (Pro)renin receptor is crucial for glioma development via the Wnt/beta-catenin signaling pathway. J. Neurosurg. 2017, 127, 819–828. [Google Scholar] [CrossRef]
- Wang, J.; Nishiyama, A.; Matsuyama, M.; Wang, Z.; Yuan, Y. The (pro)renin receptor: A novel biomarker and potential therapeutic target for various cancers. Cell Commun. Signal. 2020, 18, 39. [Google Scholar] [CrossRef]
- Nguyen, G. Renin, (pro)renin and receptor: An update. Clin. Sci. 2011, 120, 169–178. [Google Scholar] [CrossRef]
- Cruciat, C.M.; Ohkawara, B.; Acebron, S.P.; Karaulanov, E.; Reinhard, C.; Ingelfinger, D.; Boutros, M.; Niehrs, C. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science 2010, 327, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Ying, H.; Wiedemeyer, R.; Yan, H.; Quayle, S.N.; Ivanova, E.V.; Paik, J.-H.; Zhang, H.; Xiao, Y.; Perry, S.R.; et al. PLAGL2 regulates Wnt signaling to impede differentiation in neural stem cells and gliomas. Cancer Cell 2010, 17, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Pu, P.; Zhang, Z.; Kang, C.; Jiang, R.; Jia, Z.; Wang, G.; Jiang, H. Downregulation of Wnt2 and beta-catenin by siRNA suppresses malignant glioma cell growth. Cancer Gene Ther. 2009, 16, 351–361. [Google Scholar] [CrossRef]
- Pulvirenti, T.; Van Der Heijden, M.; Droms, L.A.; Huse, J.T.; Tabar, V.; Hall, A. Dishevelled 2 Signaling Promotes Self-Renewal and Tumorigenicity in Human Gliomas. Cancer Res. 2011, 71, 7280. [Google Scholar] [CrossRef] [PubMed]
- Ariza, A.; Fernandez, L.A.; Inagami, T.; Kim, J.H.; Manuelidis, E.E. Renin in glioblastoma multiforme and its role in neovascularization. Am. J. Clin. Pathol. 1988, 90, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, Y.; Jiang, Y. Podocalyxin promotes glioblastoma multiforme cell invasion and proliferation by inhibiting angiotensin-(1-7)/Mas signaling. Oncol. Rep. 2015, 33, 2583–2591. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, S.; Malla, R.; Alapati, K.; Gorantla, B.; Gujrati, M.; Dinh, D.H.; Rao, J.S. Cathepsin B and uPAR regulate self-renewal of glioma-initiating cells through GLI-regulated Sox2 and Bmi1 expression. Carcinogenesis 2013, 34, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.C.; Ding, Y.M.; Hueng, D.Y.; Chen, J.Y.; Chen, Y. Caffeine suppresses the progression of human glioblastoma via cathepsin B and MAPK signaling pathway. J. Nutr. Biochem. 2016, 33, 63–72. [Google Scholar] [CrossRef]
- Koh, S.P.; Wickremesekera, A.C.; Brasch, H.D.; Marsh, R.; Tan, S.T.; Itinteang, T. Expression of Cathepsins B, D, and G in Isocitrate Dehydrogenase-Wildtype Glioblastoma. Front. Surg. 2017, 4, 28. [Google Scholar] [CrossRef]
- Tao, W.; Chu, C.; Zhou, W.; Huang, Z.; Zhai, K.; Fang, X.; Huang, Q.; Zhang, A.; Wang, X.; Yu, X.; et al. Dual Role of WISP1 in maintaining glioma stem cells and tumor-supportive macrophages in glioblastoma. Nat. Commun. 2020, 11, 3015. [Google Scholar] [CrossRef]

