 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Raffaele Parrozzani | + 1790 word(s) | 1790 | 2021-08-12 05:33:43 | | | |
| 2 | Vivi Li | Meta information modification | 1790 | 2021-09-01 11:34:09 | | |
Video Upload Options
Type 1 neurofibromatosis (NF1) is a dominantly inherited condition predisposing to tumor development. Optic pathway glioma (OPG) is the most frequent central nervous system tumor in children with NF1, affecting approximately 15–20% of patients. The lack of well-established prognostic markers and the wide clinical variability with respect to tumor progression and visual outcome make the clinical management of these tumors challenging, with significant differences among distinct centers. We reviewed published articles on OPG diagnostic protocol, follow-up and treatment in NF1. Cohorts of NF1 children with OPG reported in the literature and patients prospectively collected in our center were analyzed with regard to clinical data, tumor anatomical site, diagnostic workflow, treatment and outcome.
1. Introduction
2. Optic Pathway Gliomas in NF1
2.1. Prevalence, Clinical Features and Natural History of OPG
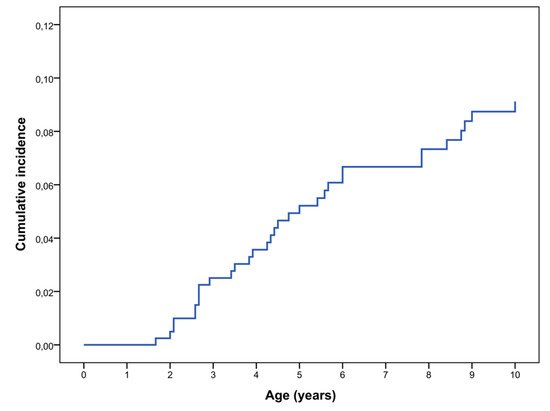
2.2. Genotype-phenotype Correlations in OPG
References
- Clementi, M.; Barbujani, G.; Turolla, L.; Tenconi, R. Neurofibromatosis-1: A maximum likelihood estimation of mutation rate. Hum. Genet. 1990, 84, 116–118.
- Xu, G.F.; Lin, B.; Tanaka, K.; Dunn, D.; Wood, D.; Gesteland, R.; White, R.; Weiss, R.; Tamanoi, F. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 1990, 63, 835–841.
- Wallis, D.; Li, K.; Lui, H.; Hu, K.; Chen, M.J.; Li, J.; Kang, J.; Das, S.; Korf, B.R.; Kesterson, R.A. Neurofibromin (NF1) genetic variant structure-function analyses using a full-length mouse cDNA. Hum. Mutat. 2018, 39, 816–821.
- Acosta, M.T.; Bearden, C.E.; Castellanos, F.X.; Castellanos, X.F.; Cutting, L.; Elgersma, Y.; Gioia, G.; Gutmann, D.H.; Lee, Y.S.; Legius, E.; et al. The Learning Disabilities Network (LeaDNet): Using neurofibromatosis type 1 (NF1) as a paradigm for translational research. Am. J. Med. Genet. A 2012, 158A, 2225–2232.
- Ferner, R.E.; Huson, S.M.; Thomas, N.; Moss, C.; Willshaw, H.; Evans, D.G.; Upadhyaya, M.; Towers, R.; Gleeson, M.; Steiger, C.; et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J. Med. Genet. 2007, 44, 81–88.
- Trevisson, E.; Cassina, M.; Opocher, E.; Vicenzi, V.; Lucchetta, M.; Parrozzani, R.; Miglionico, G.; Mardari, R.; Viscardi, E.; Midena, E.; et al. Natural history of optic pathway gliomas in a cohort of unselected patients affected by Neurofibromatosis 1. J. Neurooncol. 2017, 134, 279–287.
- Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch. Neurol. 1988, 45, 575–578.
- Sylvester, C.L.; Drohan, L.A.; Sergott, R.C. Optic-nerve gliomas, chiasmal gliomas and neurofibromatosis type 1. Curr. Opin. Ophthalmol. 2006, 17, 7–11.
- Lewis, R.A.; Gerson, L.P.; Axelson, K.A.; Riccardi, V.M.; Whitford, R.P. von Recklinghausen neurofibromatosis: II. Incidence of optic gliomata. Ophthalmology 1984, 91, 929–935.
- Listernick, R.; Charrow, J.; Greenwald, M.J.; Esterly, N.B. Optic gliomas in children with neurofibromatosis type 1. J. Pediatr. 1989, 114, 788–792.
- Listernick, R.; Charrow, J.; Greenwald, M.; Mets, M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: A longitudinal study. J. Pediatr. 1994, 125, 63–66.
- Listernick, R.; Darling, C.; Greenwald, M.; Strauss, L.; Charrow, J. Optic pathway tumors in children: The effect of neurofibromatosis type 1 on clinical manifestations and natural history. J. Pediatr. 1995, 127, 718–722.
- Listernick, R.; Louis, D.N.; Packer, R.J.; Gutmann, D.H. Optic pathway gliomas in children with neurofibromatosis 1: Consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann. Neurol. 1997, 41, 143–149.
- Listernick, R.; Charrow, J.; Gutmann, D.H. Intracranial gliomas in neurofibromatosis type 1. Am. J. Med. Genet. 1999, 89, 38–44.
- Prada, C.E.; Hufnagel, R.B.; Hummel, T.R.; Lovell, A.M.; Hopkin, R.J.; Saal, H.M.; Schorry, E.K. The Use of Magnetic Resonance Imaging Screening for Optic Pathway Gliomas in Children with Neurofibromatosis Type 1. J. Pediatr. 2015, 167, 851–856.e851.
- Blanchard, G.; Lafforgue, M.P.; Lion-François, L.; Kemlin, I.; Rodriguez, D.; Castelnau, P.; Carneiro, M.; Meyer, P.; Rivier, F.; Barbarot, S.; et al. Systematic MRI in NF1 children under six years of age for the diagnosis of optic pathway gliomas. Study and outcome of a French cohort. Eur. J. Paediatr. Neurol. 2016, 20, 275–281.
- Sellmer, L.; Farschtschi, S.; Marangoni, M.; Heran, M.K.S.; Birch, P.; Wenzel, R.; Mautner, V.F.; Friedman, J.M. Serial MRIs provide novel insight into natural history of optic pathway gliomas in patients with neurofibromatosis 1. Orphanet. J. Rare. Dis. 2018, 13, 62.
- Miller, D.T.; Freedenberg, D.; Schorry, E.; Ullrich, N.J.; Viskochil, D.; Korf, B.R.; GENETICS, C.O.; Genomics, A.C.O.M.G.A. Health Supervision for Children With Neurofibromatosis Type 1. Pediatrics 2019, 143.
- Fisher, M.J.; Loguidice, M.; Gutmann, D.H.; Listernick, R.; Ferner, R.E.; Ullrich, N.J.; Packer, R.J.; Tabori, U.; Hoffman, R.O.; Ardern-Holmes, S.L.; et al. Visual outcomes in children with neurofibromatosis type 1-associated optic pathway glioma following chemotherapy: A multicenter retrospective analysis. Neuro Oncol. 2012, 14, 790–797.
- Campen, C.J.; Gutmann, D.H. Optic Pathway Gliomas in Neurofibromatosis Type 1. J. Child. Neurol. 2018, 33, 73–81.
- Mandiwanza, T.; Kaliaperumal, C.; Khalil, A.; Sattar, M.; Crimmins, D.; Caird, J. Suprasellar pilocytic astrocytoma: One national centre’s experience. Childs. Nerv. Syst. 2014, 30, 1243–1248.
- Blazo, M.A.; Lewis, R.A.; Chintagumpala, M.M.; Frazier, M.; McCluggage, C.; Plon, S.E. Outcomes of systematic screening for optic pathway tumors in children with Neurofibromatosis Type 1. Am. J. Med. Genet. A 2004, 127A, 224–229.
- Kluwe, L.; Hagel, C.; Tatagiba, M.; Thomas, S.; Stavrou, D.; Ostertag, H.; von Deimling, A.; Mautner, V.F. Loss of NF1 alleles distinguish sporadic from NF1-associated pilocytic astrocytomas. J. Neuropathol. Exp. Neurol. 2001, 60, 917–920.
- Diggs-Andrews, K.A.; Brown, J.A.; Gianino, S.M.; Rubin, J.B.; Wozniak, D.F.; Gutmann, D.H. Sex Is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann. Neurol. 2014, 75, 309–316.
- Friedrich, R.E.; Nuding, M.A. Optic Pathway Glioma and Cerebral Focal Abnormal Signal Intensity in Patients with Neurofibromatosis Type 1: Characteristics, Treatment Choices and Follow-up in 134 Affected Individuals and a Brief Review of the Literature. Anticancer Res. 2016, 36, 4095–4121.
- Parkhurst, E.; Abboy, S. Optic Gliomas in Neurofibromatosis Type 1. J. Pediatr. Ophthalmol. Strabismus. 2016, 53, 334–338.
- Listernick, R.; Ferner, R.E.; Liu, G.T.; Gutmann, D.H. Optic pathway gliomas in neurofibromatosis-1: Controversies and recommendations. Ann. Neurol. 2007, 61, 189–198.
- Listernick, R.; Ferner, R.E.; Piersall, L.; Sharif, S.; Gutmann, D.H.; Charrow, J. Late-onset optic pathway tumors in children with neurofibromatosis 1. Neurology 2004, 63, 1944–1946.
- Créange, A.; Zeller, J.; Rostaing-Rigattieri, S.; Brugières, P.; Degos, J.D.; Revuz, J.; Wolkenstein, P. Neurological complications of neurofibromatosis type 1 in adulthood. Brain 1999, 122 Pt 3, 473–481.
- Piccirilli, M.; Lenzi, J.; Delfinis, C.; Trasimeni, G.; Salvati, M.; Raco, A. Spontaneous regression of optic pathways gliomas in three patients with neurofibromatosis type I and critical review of the literature. Childs. Nerv. Syst. 2006, 22, 1332–1337.
- Brzowski, A.E.; Bazan, C.; Mumma, J.V.; Ryan, S.G. Spontaneous regression of optic glioma in a patient with neurofibromatosis. Neurology 1992, 42, 679–681.
- Parsa, C.F.; Hoyt, C.S.; Lesser, R.L.; Weinstein, J.M.; Strother, C.M.; Muci-Mendoza, R.; Ramella, M.; Manor, R.S.; Fletcher, W.A.; Repka, M.X.; et al. Spontaneous regression of optic gliomas: Thirteen cases documented by serial neuroimaging. Arch. Ophthalmol. 2001, 119, 516–529.
- Fisher, M.J.; Avery, R.A.; Allen, J.C.; Ardern-Holmes, S.L.; Bilaniuk, L.T.; Ferner, R.E.; Gutmann, D.H.; Listernick, R.; Martin, S.; Ullrich, N.J.; et al. Functional outcome measures for NF1-associated optic pathway glioma clinical trials. Neurology 2013, 81, S15–S24.
- Taylor, T.; Jaspan, T.; Milano, G.; Gregson, R.; Parker, T.; Ritzmann, T.; Benson, C.; Walker, D.; Group, P.S. Radiological classification of optic pathway gliomas: Experience of a modified functional classification system. Br. J. Radiol. 2008, 81, 761–766.
- Guillamo, J.S.; Créange, A.; Kalifa, C.; Grill, J.; Rodriguez, D.; Doz, F.; Barbarot, S.; Zerah, M.; Sanson, M.; Bastuji-Garin, S.; et al. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): A retrospective study of 104 patients. Brain 2003, 126, 152–160.
- Fisher, M.J.; Loguidice, M.; Gutmann, D.H.; Listernick, R.; Ferner, R.E.; Ullrich, N.J.; Packer, R.J.; Tabori, U.; Hoffman, R.O.; Ardern-Holmes, S.L.; et al. Gender as a disease modifier in neurofibromatosis type 1 optic pathway glioma. Ann. Neurol. 2014, 75, 799–800.
- Balcer, L.J.; Liu, G.T.; Heller, G.; Bilaniuk, L.; Volpe, N.J.; Galetta, S.L.; Molloy, P.T.; Phillips, P.C.; Janss, A.J.; Vaughn, S.; et al. Visual loss in children with neurofibromatosis type 1 and optic pathway gliomas: Relation to tumor location by magnetic resonance imaging. Am. J. Ophthalmol. 2001, 131, 442–445.
- Zeid, J.L.; Charrow, J.; Sandu, M.; Goldman, S.; Listernick, R. Orbital optic nerve gliomas in children with neurofibromatosis type 1. J. AAPOS 2006, 10, 534–539.
- Opocher, E.; Kremer, L.C.; Da Dalt, L.; van de Wetering, M.D.; Viscardi, E.; Caron, H.N.; Perilongo, G. Prognostic factors for progression of childhood optic pathway glioma: A systematic review. Eur. J. Cancer 2006, 42, 1807–1816.
- Kehrer-Sawatzki, H.; Mautner, V.F.; Cooper, D.N. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum. Genet. 2017, 136, 349–376.
- Koczkowska, M.; Callens, T.; Gomes, A.; Sharp, A.; Chen, Y.; Hicks, A.D.; Aylsworth, A.S.; Azizi, A.A.; Basel, D.G.; Bellus, G.; et al. Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): An update of genotype-phenotype correlation. Genet. Med. 2019, 21, 867–876.
- Pinna, V.; Lanari, V.; Daniele, P.; Consoli, F.; Agolini, E.; Margiotti, K.; Bottillo, I.; Torrente, I.; Bruselles, A.; Fusilli, C.; et al. Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur. J. Hum. Genet. 2015, 23, 1068–1071.
- Rojnueangnit, K.; Xie, J.; Gomes, A.; Sharp, A.; Callens, T.; Chen, Y.; Liu, Y.; Cochran, M.; Abbott, M.A.; Atkin, J.; et al. High Incidence of Noonan Syndrome Features Including Short Stature and Pulmonic Stenosis in Patients carrying NF1 Missense Mutations Affecting p.Arg1809: Genotype-Phenotype Correlation. Hum. Mutat. 2015, 36, 1052–1063.
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-Phenotype Correlation in NF1: Evidence for a More Severe Phenotype Associated with Missense Mutations Affecting NF1 Codons 844–848. Am. J. Hum. Genet. 2018, 102, 69–87.
- Trevisson, E.; Morbidoni, V.; Forzan, M.; Daolio, C.; Fumini, V.; Parrozzani, R.; Cassina, M.; Midena, E.; Salviati, L.; Clementi, M. The Arg1038Gly missense variant in the NF1 gene causes a mild phenotype without neurofibromas. Mol. Genet. Genom. Med. 2019, 7, e616.
- Frayling, I.M.; Mautner, V.F.; van Minkelen, R.; Kallionpaa, R.A.; Aktaş, S.; Baralle, D.; Ben-Shachar, S.; Callaway, A.; Cox, H.; Eccles, D.M.; et al. Breast cancer risk in neurofibromatosis type 1 is a function of the type of. J. Med. Genet. 2019, 56, 209–219.
- Sharif, S.; Upadhyaya, M.; Ferner, R.; Majounie, E.; Shenton, A.; Baser, M.; Thakker, N.; Evans, D.G. A molecular analysis of individuals with neurofibromatosis type 1 (NF1) and optic pathway gliomas (OPGs), and an assessment of genotype-phenotype correlations. J. Med. Genet. 2011, 48, 256–260.
- Bolcekova, A.; Nemethova, M.; Zatkova, A.; Hlinkova, K.; Pozgayova, S.; Hlavata, A.; Kadasi, L.; Durovcikova, D.; Gerinec, A.; Husakova, K.; et al. Clustering of mutations in the 5’ tertile of the NF1 gene in Slovakia patients with optic pathway glioma. Neoplasma 2013, 60, 655–665.
- Hutter, S.; Piro, R.M.; Waszak, S.M.; Kehrer-Sawatzki, H.; Friedrich, R.E.; Lassaletta, A.; Witt, O.; Korbel, J.O.; Lichter, P.; Schuhmann, M.U.; et al. No correlation between NF1 mutation position and risk of optic pathway glioma in 77 unrelated NF1 patients. Hum. Genet. 2016, 135, 469–475.
- Anastasaki, C.; Morris, S.M.; Gao, F.; Gutmann, D.H. Children with 5’-end. Neurol. Genet. 2017, 3, e192.
- Xu, M.; Xiong, H.; Han, Y.; Li, C.; Mai, S.; Huang, Z.; Ai, X.; Guo, Z.; Zeng, F.; Guo, Q. Identification of Mutation Regions on. Front. Genet. 2018, 9, 270.
- Anastasaki, C.; Gao, F.; Gutmann, D.H. Commentary: Identification of Mutation Regions on. Front. Genet. 2019, 10, 115.

