 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maciej Gagat | + 2034 word(s) | 2034 | 2021-08-13 05:10:57 | | | |
| 2 | Catherine Yang | Meta information modification | 2034 | 2021-09-01 04:41:59 | | |
Video Upload Options
The transient receptor potential (TRP) melastatin-like subfamily member 2 (TRPM2) is a non-selective calcium-permeable cation channel. It is expressed by many mammalian tissues, including bone marrow, spleen, lungs, heart, liver, neutrophils, and endothelial cells. The best-known mechanism of TRPM2 activation is related to the binding of ADP-ribose to the nudix-box sequence motif (NUDT9-H) in the C-terminal domain of the channel.
1. Introduction
The transient receptor potential (TRP) melastatin-like subfamily member 2 (TRPM2; previously reported as TRPC7 or LTRPC2) is a non-selective calcium-permeable cation channel encoded by a TRPM2 gene located on the human chromosome 21 [1]. Its expression has been confirmed in many mammalian tissues, such as bone marrow, spleen, lungs, heart, liver, macrophages, neutrophils, and endothelial cells [2]. There are several isoforms of TRPM2, including the short splice variant TRPM2-S, which is a shorter form of protein devoid of 4 of 6 carboxyl-terminal transmembrane domains. The interaction between full-length TRPM2 and TRPM2-S regulates Ca 2+ influx in response to hydrogen peroxide, as confirmed in transfected 293T cells (human embryonic kidney) [3]. Because of the lacking C-terminal domain, TRPM2-S may function as an inhibitor of the full-length TRPM2 channel. Moreover, other splice variants were found in hematopoietic cells. It includes TRPM2-ΔN without amino acids 538–557 in the N-terminus, TRPM2-ΔC lacking amino acids 1292–1325 in C-terminus [4].
2. TRPM2 Structure and Activation Mechanisms
A functional form of the TRPM2 channel is a tetramer. Each monomer consists of an N-terminal region (approximately 800 amino acids in length), six transmembrane domains (S 1–S 6) with three extracellular loops, and a C-terminal domain. Both terminal domains are located inside the cell. The pore-forming loop is between S 5 and S 6. A characteristic feature of the N-terminus is the content of four TRPM subfamily homologous domains and an IQ-like motif responsible for the binding of CaM. The C-terminus contains a nudix-box sequence motif (NUDT9-H) [5]. The structure of the TRPM2 channel is shown schematically in Figure 1 .
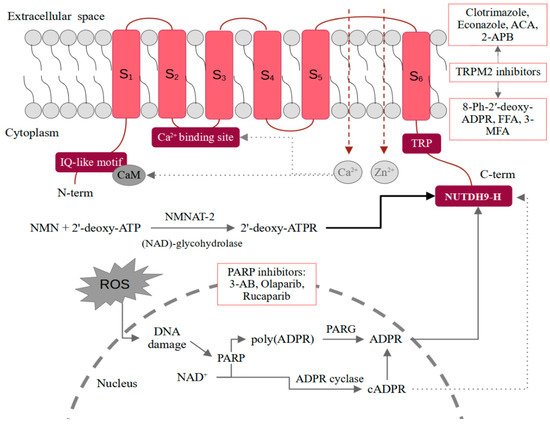
The best-known mechanism of regulation of the TRPM2 channel is the binding of ADPR to the NUDT9-H in the C-terminus [5]. The domain is also characterized by ADP hydrolase activity, which however is weak. Moreover, it seems that the prolonged interaction of ADPR with TRPM2 stimulates the channel. The intensification of enzymatic activity and the acceleration of the ADPR decomposition prevents channel opening [6]. The production of ADPR in the cells correlates with oxidative stress. Free radicals induce DNA damages, which stimulates poly-ADPR polymerase (PARP) to produce ADPR through the breakdown of NAD to poly(ADPR) and its subsequent degradation by the poly(ADPR) glycohydrolase (PARG) [7]. An alternative mechanism may be direct hydrolysis of NAD + into nicotinamide and ADPR. However, the PARP-involving mechanism seems to be much more important [2]. In the interaction of NUTDH9-H and ADPR, the ADPR terminal ribose is crucial as ADP alone does not induce channel opening [8]. An interesting issue is also the co-activation of the channel by ADPR and calcium ions. The Ca 2+ -binding site is located close to the intracellular border of the TRPM2. Only the binding of calcium ions and ADPR together is sufficient to activate the channel [9]. In general, the functioning of TRPM2 appears to be closely dependent on calcium ions. Some of the studies also suggest that the channel may be activated by the action of Ca 2+ alone [10]. An even more effective channel agonist appears to be 2′-deoxy-ADPR [11]. In vitro, it is formed from nicotinamide mononucleotide (NMN) and 2′-deoxy-ATP in a reaction catalyzed by cytosolic nicotinamide mononucleotide adenylyltransferase 2 (NMNAT-2) and nicotinamide adenine dinucleotide (NAD)-glycohydrolase CD38. As 2′-deoxy-ADPR induces 10.4-fold higher whole-cell currents compared to ADPR, it makes it a superagonist of the TRPM2 channel [11]. While in the case of ADPR and 2′-deoxy-ADPR, the situation seems quite clear, activation of TRPM2 by cADPR raises more controversy [12]. Kolisek et al. confirmed the channel opening only after administration of very high doses of cADPR. Interestingly, the simultaneous treatment with cADPR and ADPR showed a stronger effect than either of these substances alone, which may suggest their synergistic action [13]. It is also possible that cADPR is not a TRPM2 agonist at all and the effect observed in some of the studies is related to the easy degradation of cADPR to ADPR. Heiner et al. performed HPLC analysis of freshly prepared cADPR solutions and found that they contained approximately 25% ADPR. In turn, after purification, the addition of cADPR alone no longer induced intracellular ion influx in neutrophils [14]. On the contrary, Yu et al. observed effective TRPM2 activation by purified cADPR by the binding to the NUDT9-H [15]. Due to the inconsistency in the results, more reports on cADPR-dependent TRPM2 activation are necessary. It is also required to use purified cADPR and confirm the purity using, e.g., HPLC. A similar controversy is raised by the role of NAD as a TRPM2 agonist. Some reports indicate that NAD can directly activate TRPM2, while others suggest that it is due to its breakdown or contamination with ADPR [16][17][18]. Mechanisms of ligand-dependent TRPM2 activation were schematically presented in Figure 1 .
3. TRPM2 in Endothelial Cell Death
Numerous reports indicate that TRPM2 is involved in the ROS-induced death of hematopoietic cells, neurons, and vascular endothelium [19][20][21][22]. Ca 2+ entry, facilitated among others by TRPM2, leads to its rapid and excessive accumulation. In turn, disturbances in intracellular calcium homeostasis contribute to cell death. Though apoptosis is important for the maintenance of homeostasis, injury repair, and organ development, in endothelium it also facilitates pathological changes connected with inflammatory reaction and vascular diseases.
In heart microvessel endothelial cell line H5V, one of the observed effects of H 2O 2 treatment was the decrease in metabolic activity of cells observed in the MTT assay. Caspase levels examination by electrophoretic DNA fragmentation showed that the H 2O 2 caused a decrease in cell viability, which was related to apoptosis. However, the authors did not analyze the percentage of necrotic cells, which may be identified by a characteristic smear visible after the electrophoretic separation of DNA [19]. On the other hand, the use of TRPM2-specific shRNA resulted in the reduction of caspase 3, 8, and 9 activation, which indicates an inhibition of apoptosis. Additionally, it is also possible that TRPM2 is involved in the induction of cell death by one of the proinflammatory cytokines tumor necrosis factor alpha (TNFα). 36 h treatment of H5V cells with 10 ng/mL of TNFα resulted in a decrease in cell viability. This effect was limited after using TRPM2 blocking antibody (TM2E3) or TRPM2-specific shRNA [19].
Furthermore, TRPM2 may be involved in high glucose-induced mitochondrial fission in endothelial cells [23]. The process of mitochondrial fission is strictly connected with apoptosis [24]. One of the consequences of diabetes is a high concentration of circulating glucose. It leads to the production of ROS, which affects endothelial cells and whole circulatory system. Abuarab et al. showed that inhibition of TRPM2 using its inhibitor or TRPM2 silencing RNA blocked the mitochondrial fission in HUVEC cells [23]. Similarly, primary endothelial cells obtained from TRPM2 knockout mice were less susceptible to high glucose-induced mitochondrial fission compared to wild-type mice. Moreover, the observed effect was not connected with store-operated Ca 2+ entry as the silencing of stromal interaction molecule 1 and Orai-1 did not affect the high glucose-induced calcium influx. The authors proved that the observed effect was connected with ROS production stimulated by high glucose concentration as preincubation with N -acetyl-cysteine (ROS scavenger) prevented mitochondrial fragmentation [23].
It is also worth mentioning that TRPM2 may contribute to the type II programmed cell death called autophagy. Although no reports are showing TRPM2 involvement in autophagy induction in endothelial cells, there is evidence of its participation in this type of cell death in vascular smooth muscles and brain pericyte injury [25][26]. In primary cultured mouse aortic smooth muscle cells (mASMCs) TRPM2 expression was found in lysosomes/autolysosomes resulting from nutrients starvation. Both autophagic response and lysosomal /autolysosomal acidification were reduced after TRPM2 knockout [25]. In turn, in the human brain vascular pericytes, zinc oxide nanoparticles (ZnO-NP)-induced autophagy was accompanied by increased TRPM2-S expression. Knockout of TRPM2 preserved the structure of brain microvessels in mice and prevented vascular injury induced by ZnO-NP [26]. Thus, the TRPM2-dependent influx of calcium ions appears to have a broad effect on the circulatory system. However, in the case of autophagy, the matter remains controversial as it can be cytoprotective or act as a cell death mechanism [27].
4. TRPM2 Inhibitors in Clinical Application and Further Perspectives
Although numerous TRPM2 inhibitors are known, they are unable to target the TRPM2 channel with appropriate specificity. The examples are N-(p-Amylcinnamoyl) anthranilic acid, widely used in research, which additionally affects TRPM8, TRPC6, TPPV1, TRPC3, or flufenamic acid inhibiting TRPC3, TRPC7, TRPM2–5, TRPM7/8, TRPV1, TRPV3/4 [28]. However, some efforts are made to enhance the specificity of ACA by using its derivatives [29]. Moreover, some of the substances are characterized by low inhibitory effects or poor membrane permeabilization properties [30]. In turn, some of the promising inhibitors lacking stability in in vivo conditions [31]. Recently, the influence of various substances of natural origin, such as resveratrol or curcumin in the context of TRPM2 inhibition, has been intensively studied [32][33]. However, so far, no similar studies have been performed on endothelial cells. The substances of natural origin will likely encounter the same problems as other inhibitors, i.e., lack of specificity and stability [34]. Some of the potential inhibitors of TRPM2 are included in Figure 1 .
Since the opening of the TRPM2 channel is observed in the course of increased oxidative stress, the logical approach seems to be the use of antioxidants. However, in this case, the results remain inconclusive. As showed by Nazıroğlu and Lückhoff, treatment of Chinese hamster ovary (CHO) cells with antioxidants such as ascorbic acid (vitamin C), alpha-tocopherol (vitamin E), or glutathione did not prevent or even weaken the H 2O 2-induced TRPM2 opening and following Ca 2+ current [35]. However, this study has several limitations, some of which have also been described by the authors. The first problem is the optimization stage of the study. The doses of the antioxidants selected by the researchers could be inadequate. The concentration of hydrogen peroxide used in the study is also significant here. Due to the inability to activate TRPM2 with low concentrations of H 2O 2, mM concentrations were used in the study. For most cells, severe changes are induced already with nM or µM oxidant concentrations. In this case, the doses of antioxidants should also be increased accordingly. Moreover, later in the study, cells transfected with the expression plasmid were used to increase TRPM2 expression. The obtained results are therefore of little importance in the clinical context. It might be more valuable to use cells that naturally are characterized by a relatively high TRPM2 expression in similar research. An important variable here is also the experiment design and treatment regimen. Whether and how long cells should be pretreated with the selected compounds, or maybe a better solution would be the simultaneous administration of an antioxidant and a substance that intensifies oxidative stress. As endothelial cells naturally express TRPM2-S, it may turn out to be the safest method. However, currently, no protocols allow effective TRPM2-S upregulation and can be used in the clinical context.
Another option is inhibition of PARP, which prevents the formation of TRPM2 which prevents the formation of ADPR as an activator of TRPM2 channels. Although some PARP inhibitors are already approved by Food and Drug Administration (FDA) and European Medicine Agency (EMA), they are currently used mostly in cancer treatment. However, there is strong evidence suggesting that PARP inhibitors improve endothelial function impaired by the aging process [36][37][38]. However, due to the involvement of PARP in such important and complex processes as DNA damage repair, the observed effect of the inhibitors will not be at all or will be only partially related to the blockade of ADPR-induced TRPM2 activation. Chosen PARP inhibitors were included in Figure 1 .
References
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-Permeable Channel Activated by Changes in Redox Status Confers Susceptibility to Cell Death. Mol. Cell 2002, 9, 163–173.
- Hecquet, C.M.; Ahmmed, G.U.; Vogel, S.M.; Malik, A.B. Role of TRPM2 Channel in Mediating H2O2 -Induced Ca2+ Entry and Endothelial Hyperpermeability. Circ. Res. 2008, 102, 347–355.
- Zhang, W.; Chu, X.; Tong, Q.; Cheung, J.Y.; Conrad, K.; Masker, K.; Miller, B.A. A Novel TRPM2 Isoform Inhibits Calcium Influx and Susceptibility to Cell Death. J. Biol. Chem. 2003, 278, 16222–16229.
- Wehage, E.; Eisfeld, J.; Heiner, I.; Jüngling, E.; Zitt, C.; Lückhoff, A. Activation of the Cation Channel Long Transient Receptor Potential Channel 2 (LTRPC2) by Hydrogen Peroxide. J. Biol. Chem. 2002, 277, 23150–23156.
- Kühn, F.J.P.; Lückhoff, A. Sites of the NUDT9-H Domain Critical for ADP-ribose Activation of the Cation Channel TRPM2. J. Biol. Chem. 2004, 279, 46431–46437.
- Perraud, A.-L.; Shen, B.; Dunn, C.A.; Rippe, K.; Smith, M.K.; Bessman, M.J.; Stoddard, B.L.; Scharenberg, A.M. NUDT9, a Member of the Nudix Hydrolase Family, Is an Evolutionarily Conserved Mitochondrial ADP-ribose Pyrophosphatase. J. Biol. Chem. 2003, 278, 1794–1801.
- Harrision, D.; Gravells, P.; Thompson, R.; Bryant, H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP) – Function in Genome Maintenance and Relevance of Inhibitors for Anti-cancer Therapy. Front. Mol. Biosci. 2020, 7.
- Fliegert, R.; Watt, J.M.; Schöbel, A.; Rozewitz, M.D.; Moreau, C.; Kirchberger, T.; Thomas, M.P.; Sick, W.; Araujo, A.C.; Harneit, A.; et al. Ligand-induced activation of human TRPM2 requires the terminal ribose of ADPR and involves Arg1433 and Tyr1349. Biochem. J. 2017, 474, 2159–2175.
- Wang, L.; Fu, T.-M.; Zhou, Y.; Xia, S.; Greka, A.; Wu, H. Structures and gating mechanism of human TRPM2. Science 2018, 362.
- Luo, Y.-H.; Yu, X.-F.; Ma, C.; Yang, F.; Yang, W. Effects of calcium-binding sites in the S2–S3 loop on human and Nematostella vectensis TRPM2 channel gating processes. J. Zhejiang Univ. Sci. B 2019, 20, 972–982.
- Fliegert, R.; Bauche, A.; Pérez, A.-M.W.; Watt, J.M.; Rozewitz, M.D.; Winzer, R.; Janus, M.; Gu, F.; Rosche, A.; Harneit, A.; et al. 2′-Deoxyadenosine 5′-diphosphoribose is an endogenous TRPM2 superagonist. Nat. Chem. Biol. 2017, 13, 1036–1044.
- Fliegert, R.; Riekehr, W.M.; Guse, A.H. Does Cyclic ADP-Ribose (cADPR) Activate the Non-selective Cation Channel TRPM2? Front. Immunol. 2020, 11.
- Kolisek, M.; Beck, A.; Fleig, A.; Penner, R. Cyclic ADP-Ribose and Hydrogen Peroxide Synergize with ADP-Ribose in the Activation of TRPM2 Channels. Mol. Cell 2005, 18, 61–69.
- Heiner, I.; Eisfeld, J.; Warnstedt, M.U.; Radukina, N.; Jüngling, E.; Lückhoff, A. Endogenous ADP-ribose enables calcium-regulated cation currents through TRPM2 channels in neutrophil granulocytes. Biochem. J. 2006, 398, 225–232.
- Yu, P.; Liu, Z.; Yu, X.; Ye, P.; Liu, H.; Xue, X.; Yang, L.; Li, Z.; Wu, Y.; Fang, C.; et al. Direct Gating of the TRPM2 Channel by cADPR via Specific Interactions with the ADPR Binding Pocket. Cell Rep. 2019, 27, 3684–3695.e4.
- Tóth, B.; Iordanov, I.; Csanády, L. Ruling out pyridine dinucleotides as true TRPM2 channel activators reveals novel direct agonist ADP-ribose-2′-phosphate. J. Gen. Physiol. 2015, 145, 419–430.
- Huang, Y.; Roth, B.; Lü, W.; Du, J. Ligand recognition and gating mechanism through three ligand-binding sites of human TRPM2 channel. eLife 2019, 8.
- Huang, Y.; Winkler, P.A.; Sun, W.; Lü, W.; Du, J. Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium. Nat. Cell Biol. 2018, 562, 145–149.
- Sun, L.; Yau, H.-Y.; Wong, W.-Y.; Li, R.A.; Huang, Y.; Yao, X. Role of TRPM2 in H2O2-Induced Cell Apoptosis in Endothelial Cells. PLoS ONE 2012, 7, e43186.
- Hecquet, C.M.; Zhang, M.; Mittal, M.; Vogel, S.M.; Di, A.; Gao, X.; Bonini, M.G.; Malik, A.B. Cooperative Interaction oftrpMelastatin Channel Transient Receptor Potential (TRPM2) With Its Splice Variant TRPM2 Short Variant Is Essential for Endothelial Cell Apoptosis. Circ. Res. 2014, 114, 469–479.
- Osmanlıoğlu, H. Ömer; Yıldırım, M.K.; Akyuva, Y.; Yıldızhan, K.; Nazıroğlu, M. Morphine Induces Apoptosis, Inflammation, and Mitochondrial Oxidative Stress via Activation of TRPM2 Channel and Nitric Oxide Signaling Pathways in the Hippocampus. Mol. Neurobiol. 2020, 57, 3376–3389.
- Zhang, W.; Hirschler-Laszkiewicz, I.; Tong, Q.; Conrad, K.; Sun, S.-C.; Penn, L.; Barber, D.L.; Stahl, R.; Carey, D.J.; Cheung, J.Y.; et al. TRPM2 is an ion channel that modulates hematopoietic cell death through activation of caspases and PARP cleavage. Am. J. Physiol. Cell Physiol. 2006, 290, C1146–C1159.
- Abuarab, N.; Munsey, T.S.; Jiang, L.-H.; Lin-Hua, J.; Sivaprasadarao, A. High glucose–induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn2+-mediated mitochondrial fission. Sci. Signal. 2017, 10.
- Youle, R.J.; Karbowski, M. Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 657–663.
- Zhao, Q.; Li, J.; Ko, W.-H.; Kwan, Y.-W.; Jiang, L.; Sun, L.; Yao, X. TRPM2 promotes autophagic degradation in vascular smooth muscle cells. Sci. Rep. 2020, 10, 1–11.
- Jiang, Q.; Gao, Y.; Wang, C.; Tao, R.; Wu, Y.; Zhan, K.; Liao, M.; Lu, N.; Lu, Y.; Wilcox, C.S.; et al. Nitration of TRPM2 as a Molecular Switch Induces Autophagy During Brain Pericyte Injury. Antioxid. Redox Signal. 2017, 27, 1297–1316.
- Izdebska, M.; Zielińska, W.; Hałas-Wiśniewska, M.; Grzanka, A. Involvement of Actin in Autophagy and Autophagy-Dependent Multidrug Resistance in Cancer. Cancers 2019, 11, 1209.
- Zhang, H.; Zhao, S.; Yu, J.; Yang, W.; Liu, Z.; Zhang, L. Medicinal chemistry perspective of TRPM2 channel inhibitors: Where we are and where we might be heading? Drug Discov. Today 2020, 25, 2326–2334.
- Zhang, H.; Yu, P.; Lin, H.; Jin, Z.; Zhao, S.; Zhang, Y.; Xu, Q.; Liu, Z.; Yang, W.; Zhang, L. The Discovery of Novel ACA Derivatives as Specific TRPM2 Inhibitors that Reduce Ischemic Injury Both In Vitro and In Vivo. J. Med. Chem. 2021, 64, 3976–3996.
- Luo, X.; Lihe, Z.; Zhan, K.; Yang, W.; Zhang, L.; Wang, K.; Kaiyu, Z.; Zhang, L. Selective inhibition of TRPM2 channel by two novel synthesized ADPR analogues. Chem. Biol. Drug Des. 2018, 91, 552–566.
- Fourgeaud, L.; Dvorak, C.; Faouzi, M.; Starkus, J.; Sahdeo, S.; Wang, Q.; Lord, B.; Coate, H.; Taylor, N.; He, Y.; et al. Pharmacology of JNJ-28583113: A novel TRPM2 antagonist. Eur. J. Pharmacol. 2019, 853, 299–307.
- Akyuva, Y.; Nazıroğlu, M. Resveratrol attenuates hypoxia-induced neuronal cell death, inflammation and mitochondrial oxidative stress by modulation of TRPM2 channel. Sci. Rep. 2020, 10, 1–16.
- Özkaya, D.; Nazıroğlu, M. Curcumin diminishes cisplatin-induced apoptosis and mitochondrial oxidative stress through inhibition of TRPM2 channel signaling pathway in mouse optic nerve. J. Recept. Signal Transduct. 2020, 40, 97–108.
- Lin, Y.; Chen, F.; Zhang, J.; Wang, T.; Wei, X.; Wu, J.; Feng, Y.; Dai, Z.; Wu, Q. Neuroprotective Effect of Resveratrol on Ischemia/Reperfusion Injury in Rats Through TRPC6/CREB Pathways. J. Mol. Neurosci. 2013, 50, 504–513.
- Nazıroğlu, M.; Lückhoff, A. Effects of antioxidants on calcium influx through TRPM2 channels in transfected cells activated by hydrogen peroxide. J. Neurol. Sci. 2008, 270, 152–158.
- Radovits, T.; Zotkina, J.; Lin, L.-N.; Bömicke, T.; Arif, R.; Gerö, D.; Horvath, E.M.; Karck, M.; Szabó, C.; Szabó, G. Poly(ADP-Ribose) Polymerase Inhibition Improves Endothelial Dysfunction Induced by Hypochlorite. Exp. Biol. Med. 2007, 232, 1204–1212.
- Tarantini, S.; Yabluchanskiy, A.; Csipo, T.; Fulop, G.; Kiss, T.; Balasubramanian, P.; DelFavero, J.; Ahire, C.; Ungvari, A.; Nyúl-Tóth, Ádám; et al. Treatment with the poly(ADP-ribose) polymerase inhibitor PJ-34 improves cerebromicrovascular endothelial function, neurovascular coupling responses and cognitive performance in aged mice, supporting the NAD+ depletion hypothesis of neurovascular aging. GeroScience 2019, 41, 533–542.
- Zhang, G.-H.; Chao, M.; Hui, L.-H.; Xu, D.L.; Cai, W.-L.; Zheng, J.; Gao, M.; Zhang, M.-X.; Wang, J.; Lu, Q.-H. Poly(ADP-ribose)Polymerase 1 Inhibition Protects Against Age-Dependent Endothelial Dysfunction. Clin. Exp. Pharmacol. Physiol. 2015, 42, 1266–1274.

