 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mario Vitale | + 2107 word(s) | 2107 | 2021-08-04 07:51:53 | | | |
| 2 | Ron Wang | + 330 word(s) | 2437 | 2021-08-30 10:21:30 | | | | |
| 3 | Ron Wang | + 273 word(s) | 2380 | 2021-08-30 10:21:54 | | |
Video Upload Options
The significance of RAS mutations as a prognostic marker has changed in recent years. At the beginning, RAS mutations were considered a promoter of progressive dedifferentiation in benign nodules, and as such a risk factor for evolution from adenoma to carcinoma, therefore a marker of poorer prognosis in DTC. It is clear now that the impact of RAS mutations on thyroid cell, normal and transformed, and the impact on the clinical behavior of thyroid neoplasms must not to be considered by itself but rather together with other genetic abnormalities in a more complex contest.
1. Introduction
The majority of thyroid nodules are benign and require only clinical and ultrasonographic follow up [1]. In few cases it is a cancer whose clinical behavior can be extremely indolent or sometime aggressive. Fine needle-aspiration cytology (FNAC) is the gold standard for diagnosis [2]. Unfortunately, 5–10% of FNAC are not diagnostic because of inadequate sampling and 20% to 25% do not clearly indicate the nature of the lesion, so that these nodules are classified as indeterminate, posing difficulties for management [3][4].
Testing for genetic mutations has been proposed for indeterminate cytology to improve FNAC accuracy and for prognostic purpose, supporting the clinicians’ decision making process. Mutations of rat sarcoma viral oncogene homolog (RAS), rearranged during transfection/PTC (RET/PTC) , v-Raf murine sarcoma viral oncogene homolog B1 (BRAF), telomerase reverse transcriptase (TERT), and rearrangement of paired box gene 8-peroxisome /proliferator-activator receptor γ (Pax8/PPARγ), are some of the genetic mutations useful to improve the diagnostic accuracy of needle biopsy when added to cytological evaluation. For this reason, mutational analysis is recommended in the last revisions of the guidelines for the diagnosis and management of thyroid nodules with indeterminate cytology [5][6]. The most popular molecular test used is the seven-gene panel of mutations (including RAS, BRAF, RET/PTC, and Pax8/PPARγ) with an estimated sensitivity of 57–75%, specificity of 97–100%, PPV of 87–100%, and NPV of 79–86% [7][8][9].
Although RAS mutations are included in these tests, they have low diagnostic sensitivity and specificity by themselves, benign nodules harboring RAS mutations have uncertain malignant potential, and the role of mutated RAS in the clinical outcomes of thyroid cancer is uncertain. This scenario has been further complicated by the recent introduction of a new histology category of encapsulated follicular variant of papillary thyroid carcinoma, with highly indolent behavior. For all these reasons, the significance of RAS mutations and how to appropriately use them to assist the management of thyroid nodules and thyroid cancer is unclear.
2. Oncogenicity of Ras Mutants
RAS mutants have been found in 95% of pancreatic ductal adenocarcinomas, 50% of colon cancers, and 30% of non–small cell lung cancers (NSCLC). Overall, activating mutations in RAS are found in 32% of human cancers ( https://cancer.sanger.ac.uk/cosmic , accessed on 9 June 2021).
The role of Ras in thyroid cell differentiation and tumorigenesis has been demonstrated in animal experimental models ( Figure 1 ). Only 10% percent of rats whose thyroid was injected with a retrovirus carrying mutant KRAS developed thyroid carcinomas of very small size and with a one-year latency. Only the treatment of the animals with a goitrogenic agent induced thyroid carcinomas within three months in 90% of the animals. Transgenic mice bearing mutated KRAS under the control of rat thyroglobulin promoter developed only thyroid adenomas at low frequency and long latency, while the frequency increased with a concomitant treatment with a goitrogenic agent as occurred in retrovirus-injected animals [10]. These results demonstrate a low oncogenic potential of KRas mutants in the thyroid, boosted by a cofactor as high TSH stimulation. Similar results with the development of rare papillary thyroid carcinomas were obtained with mutant HRas under the control of bovine thyroglobulin promoter [11]. These results suggest that HRas and KRas mutations alone are not sufficient to induce thyroid carcinomas, while they could act as a predisposing factor. According to this hypothesis is the observation that isolated expression of HRasG12V or loss of NF2 are insufficient to drive thyroid tumorigenesis, while their coexpression in the thyroid induces large thyroid cancers, mostly poorly differentiated, with high penetrance [12]. One more example of cooperation among RAS mutants and other effectors was proposed by Krishnamoorthy et al. [13]. Mice expressing the eukaryotic initiation factor 1A hotspot splice-site mutation (EIF1AX-A113splice) developed only thyrocytes hyperplasia with atypical features, whereas mice coexpressing EIF1AX-A113splice and HRasG12V developed Hurthle cell adenomas, papillary thyroid carcinomas (PTC), and poorly differentiated thyroid cancer (PDTC) with high penetrance and close phenocopy of human thyroid tumors harboring the combined genetic lesions [14].
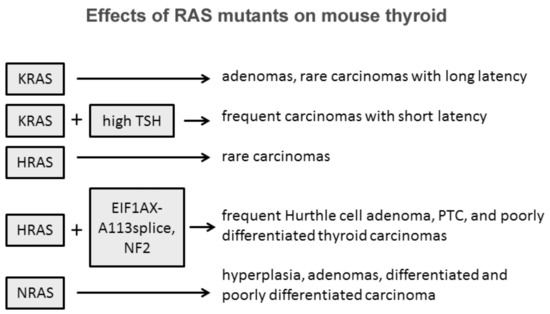
Some evidences suggest that among the three isoforms, NRAS mutations are those more oncogenic in the thyroid. Transgenic mice expressing mutated human NRAS G61 specifically in thyroid follicular cells developed progressive lesions from follicular cell hyperplasia to adenoma and carcinoma [15]. Thirty percent of mice developed differentiated tumors with follicular or mixed papillary-follicular malignant features and 10% of mice developed invasive carcinomas with large poorly differentiated areas. Furthermore, distant metastases were observed in the liver of three mice, in the lung of two mice, and in the femur of one mouse. This striking difference between RAS isoforms might be explained with the intrinsic tissue-specific different isoform effects and role of members of the RAS family as suggested by transgenic mice experimental models where RAS expression was not tissue-restricted [16][17][18][19].
Overall, these experimental models demonstrate that RAS overexpression and RAS mutants represent a favorable genetic ground in thyroid follicular cells and that the collaboration of other effectors and/or genetic events is necessary in conferring the cancer phenotype. Among RAS isoforms, NRAS displays a stronger albeit limited oncogenic potential, a conclusion supported also by studies in spontaneous thyroid tumors.
3. Prevalence of RAS Mutations in Thyroid Nodular Disease
Thyroid cancer is among the first tumors where activating RAS mutations were recognized [20][21]. Quite soon it appeared clear that RAS mutations were detectable at all stages of thyroid tumorigenesis, being found in 33% of adenomas, 53% of differentiated follicular carcinomas and 60% of undifferentiated carcinomas [22].
Although there is a general agreement that RAS mutations are present at any stage of thyroid oncogenesis, the range of prevalence is quite broad as a consequence of pathology classification and the sensitivity of detection method applied. In early studies, polymerase chain reaction-single-strand conformational polymorphism (SSCP), in many cases followed by DNA sequencing, was the most frequently used method for the detection of RAS mutations. This method yielded a lower prevalence with respect to sequencing in all histotypes but FTC, revealing the presence of an RAS mutation in about 2.3% of nodular hyperplasia (NH) (1/43), 13% of follicular adenomas (FAD) (13/103), 10% of PTC (16/165) and 26% of follicular thyroid carcinomas (FTC) (11/43) [23][24][25][26][27][28][29]. Direct sequencing of PCR products resulted in a higher percentage of RAS mutations: 9.3% in NH (11/115), 22% in FAD (56/257), 23% in PTC (115/508) and 29% in FTC (36/125) [30][31][32][33][34][35][36][37][38][39].
Many studies on surgical samples with a definitive diagnosis have been performed to assess the prevalence of all RAS mutations in benign and malignant thyroid nodules ( Table 1 ). In 2296 tissue samples, NRAS is the isoform most frequently mutated (59.4% of all RAS mutations), and among all activating point mutations NRAS61 is the most frequent (56.9%). The prevalence of RAS mutation in tissue samples of NH, FAD, FTC, and PTC were, respectively, 8.5%, 21.0%, 29.8%, and 11.5%.
| Study | Year | Method | Tumor Histotype | N | HRAS | KRAS | NRAS | All RAS |
All RAS % |
|||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 12/13 | 61 | 12/13 | 61 | 12/13 | 61 | |||||||
| Lemoine, N.R. [24] | 1989 | SB | FAD | 24 | 1 | 2 | 1 | 0 | 1 | 3 | 8 | 33.3 |
| FTC | 10 | 1 | 2 | 0 | 0 | 0 | 1 | 4 | 40 | |||
| Namba, H. [40] | 1990 | SB | PTC | 14 | 3 | 0 | 0 | 0 | 0 | 0 | 3 | 21.4 |
| FTC | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |||
| NH | 19 | 4 | 0 | 0 | 0 | 0 | 0 | 4 | 21.1 | |||
| FAD | 24 | 4 | 0 | 0 | 0 | 0 | 2 | 6 | 25 | |||
| Shi, Y. [41] | 1991 | SB | FTC | 16 | 0 | 3 | 0 | 0 | 0 | 1 | 4 | 25 |
| PTC | 22 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |||
| FAD | 25 | 0 | 12 | 0 | 0 | 0 | 2 | 14 | 56 | |||
| Yoshimoto, K. [28] | 1992 | SSCP | FAD | 24 | 0 | 1 | 0 | 0 | 0 | 4 | 5 | 20.8 |
| PTC | 26 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |||
| Manenti, G. [36] | 1994 | SEQ | FTC | 19 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Ezzat, S. [25] | 1996 | SSCP | NH | 18 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| FAD | 7 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 14.3 | |||
| PTC | 16 | 1 | 0 | 0 | 0 | 0 | 1 | 2 | 12.5 | |||
| FTC | 4 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 25 | |||
| Naito, H. [29] | 1998 | SSCP | PTC | 34 | 0 | 0 | 5 | 1 | 0 | 1 | 7 | 20.6 |
| Bouras, M. [34] | 1998 | SEQ | FAD | 35 | 0 | 22.9 | ||||||
| PTC | 66 | 0 | 9.1 | |||||||||
| FTC | 6 | 0 | 16.7 | |||||||||
| Sugg, S.L. [26] | 1999 | SSCP | PTC | 20 | 1 | 1 | 2 | 10 | ||||
| Esapa, C.T. [31] | 1999 | SSCP | NH | 25 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 4 |
| FAD | 41 | 0 | 0 | 0 | 2 | 0 | 5 | 7 | 18.4 | |||
| FTC | 8 | 0 | 0 | 0 | 0 | 0 | 4 | 4 | 50 | |||
| PTC | 12 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 8.3 | |||
| PTC | 46 | 0 | 0 | 0 | 0 | |||||||
| Motoi, N. [32] | 2000 | SEQ | FAD | 5 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | 40 |
| PTC | 16 | 1 | 0 | 3 | 0 | 0 | 7 | 11 | 68.8 | |||
| FTC | 4 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 25 | |||
| Nikiforova, M.N. [42] | 2003 | FMCA | FAD | 23 | 2 | 6 | 8 | 34.8 | ||||
| FTC | 33 | 2 | 12 | 14 | 42.4 | |||||||
| FTC | 19 | 1 | 1 | 2 | 10.5 | |||||||
| Vasko, V. [33] | 2003 | SEQ | FAD | 46 | 0 | 0 | 0 | 0 | 0 | 7 | 7 | 15.2 |
| FTC | 34 | 1 | 0 | 0 | 0 | 0 | 6 | 7 | 20.6 | |||
| Soares, P. [27] | 2003 | SSCP | FAD | 34 | 0 | 14.7 | ||||||
| PTC | 27 | 0 | 7.4 | |||||||||
| FTC | 12 | 0 | 33.3 | |||||||||
| Garcia-Rostan, G. [43] | 2003 | SSCP | PTC | 30 | 0 | 6.7 | ||||||
| FTC | 19 | 0 | 10.5 | |||||||||
| Zhu, Z. [44] | 2003 | PCR | PTC FV | 30 | 3 | 10 | 13 | 43.3 | ||||
| Liu, R.T. [35] | 2004 | SEQ | NH | 20 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| FAD | 17 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 5.9 | |||
| PTC | 42 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |||
| FTC | 8 | 0 | 1 | 0 | 1 | 0 | 1 | 3 | 37.5 | |||
| Fukahori, M. [45] | 2012 | LHSA | FAD | 40 | 0 | 4 | 0 | 0 | 4 | 4 | 12 | 30 |
| FTC | 58 | 2 | 3 | 3 | 2 | 1 | 22 | 33 | 56.9 | |||
| Park, J.Y. [38] | 2013 | SEQ | PTC FV | 132 | 0 | 4 | 0 | 8 | 1 | 22 | 35 | 26.5 |
| Schulten, H.J. [46] | 2013 | SEQ | NH | 18 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 5.6 |
| FAD | 69 | 0 | 5 | 0 | 0 | 0 | 10 | 15 | 21.7 | |||
| PTC | 89 | 0 | 4 | 0 | 1 | 0 | 7 | 12 | 13.5 | |||
| FTC | 17 | 0 | 1 | 0 | 0 | 0 | 1 | 2 | 11.8 | |||
| Giordano, T.J. [37] | 2014 | SEQ | NH | 27 | 0 | 3.7 | ||||||
| FAD | 28 | 0 | 17.9 | |||||||||
| PTC | 36 | 0 | 13.9 | |||||||||
| FTC | 17 | 0 | 17.6 | |||||||||
| McFadden, D.G. [39] | 2014 | SNS | PTC FV | 127 | 0 | 36.2 | ||||||
| Giordano, T.J. [37] | 2014 | NGS | PTC | 403 | 14 | 2 | 1 | 34 | 51 | 12.9 | ||
| Jeong, S.H. [47] | 2018 | PSEQ | NH | 50 | 0 | 3 | 6 | 9 | 18 | |||
| FAD | 57 | 0 | 5 | 5 | 1 | 1 | 10 | 22 | 31.6 | |||
| FTC | 39 | 0 | 4 | 6 | 3 | 1 | 7 | 21 | 48.7 | |||
| Duan, H. [48] | 2019 | NGS | FTC | 10 | 1 | 2 | 0 | 0 | 0 | 1 | 4 | 40 |
| FAD | 48 | 4 | 3 | 7 | 14.6 | |||||||
| Lemoine, N.R. [24] | 1989 | SB | FAD | 24 | 1 | 2 | 1 | 0 | 1 | 3 | 8 | 33.3 |
| FTC | 10 | 1 | 2 | 0 | 0 | 0 | 1 | 4 | 40 | |||
| Namba, H. [40] | 1990 | SB | PTC | 14 | 3 | 0 | 0 | 0 | 0 | 0 | 3 | 21.4 |
In the context of differentiated thyroid cancer (DTC), representing almost the totality of follicular-cell derived thyroid malignancies [46], the detection of RAS mutations is related to the follicular architecture. Indeed, mutated RAS is not only present in a relevant portion of FTC, but among PTCs, it is more frequently detected in the follicular variant (FVPTC) [47][40], characterized by a nearly total follicular pattern coupled with cytological PTC features. Actually, RAS mutations are typically present in the encapsulated FVPTC (EFVPTC), whereas the unencapsulated/infiltrative FVPTC (IFVPTC) frequently carry the BRAFV600E mutation and are characterized by more aggressive behavior [41][42].
4. Role of RAS Mutations in the Prognosis of Benign and Malignant Thyroid Nodules
Although the number of studies on the evolution of benign nodules harboring RAS mutations is still very limited for patient numerosity and follow-up duration, the stability observed so far challenges the hypothesis that RAS mutation is a risk factor for malignant transformation. The natural history of cytologically benign thyroid nodules with unknown mutational status argues against the role of RAS mutations as a risk factor for thyroid cancer. In a retrospective study on 330 cytologically benign nodules followed for a mean of 20 months (range 1 month–5 years), many of them re-evaluated by a second FNAC, only one was demonstrated to be a PDTC [43]. One hundred benign solid nodules did not display signs of risk for thyroid cancer over a 9 to 11 years follow-up and of 21 nodules re-examined by FNAC only one case turned out to be malignant [44]. In a prospective study involving 992 consecutive patients with cytologically benign thyroid nodules of unknown RAS mutational status, only five nodules proved to be malignant after 5 years of follow-up, that is, only 0.3% of all nodules [45]. The spare reclassified cases are compatible with a misdiagnosis and are much lower than the number of RAS mutation-positive nodules expected, suggesting that RAS mutation is not a risk factor for the malignant evolution of benign nodules. To date, in the absence of evidence that cytologically benign nodules bearing RAS mutations evolve toward malignancy or have a different progression, testing the RAS mutational status is not recommended. A limit of these studies is that the majority of them have been performed in nodules with benign cytology and these conclusions cannot be automatically extended to nodules with indeterminate cytology, where other genetic mutations can coexist with RAS mutations.
Prognostic studies of RAS mutational status in thyroid cancer generated conflicting results. Earlier studies have reported the association between RAS mutations and a more aggressive phenotype of thyroid cancer [48]. In a series of 88 carcinomas, an NRAS mutation was present in 13.9% of well-differentiated carcinomas (WDTC), in 18.2% of PDTC and 37.5% of undifferentiated carcinomas [49]. In the study by Garcia-Rostan et al. a RAS mutation was found in 8.2% of WDTC, in 55.2% PDTC, and in 51.7% of undifferentiated or anaplastic carcinomas (UTC) [50]. A cumulative analysis of 834 thyroid cancers including 492 WDTC, 173 PDTC and 169 UTC thyroid cancers, displayed a RAS mutation in 151 tumors ( Table 2 ). RAS mutations were more frequent in PDTC (27.7%) then in WDTC (14.8%) and UTC (17.7%), confirming the hypothesis that RAS is involved in thyroid cancer progression or it is simply associated with it, whereas the undifferentiated/anaplastic thyroid carcinoma does not necessarily arise from a WDTC and it is more linked to other genetic alterations. However, the dilemma whether de-differentiation of WDTC is accompanied by RAS mutations or RAS promotes the de-differentiation in spontaneous thyroid cancer is unsolved.
| Year | WDTC n/Total |
WDTC % |
PDTC n/Total |
PDTC % |
UTC n/Tot |
UTC % |
|
|---|---|---|---|---|---|---|---|
| Lemoine, N.R. [24] | 1989 | 8/15 | 53.3 | 6/10 | 60.0 | - | - |
| Suarez, H.G. [22] | 1988 | 8/13 | 61.5 | 1/10 | 10.0 | - | - |
| Du Villard, J.A. [51] | 1995 | 1/6 | 16.7 | 6/14 | 42.8 | 1/1 | 100 |
| Bouras, M. [34] | 1998 | 6/66 | 9.0 | - | - | 0/8 | 0.0 |
| Ricarte-Filho, J.C. [52] | 2009 | 1/11 | 9.0 | 8/55 | 14.5 | 0/2 | 0.0 |
| Liu, Z. [53] | 2008 | 13/64 | 20.3 | - | - | 4/51 | 7.8 |
| Santarpia, L. [54] | 2008 | 0/8 | 0.0 | - | - | 2/15 | 13.3 |
| Manenti, G. [36] | 1994 | 5/52 | 9.6 | 3/11 | 27.3 | 1/5 | 20.0 |
| Garcia-Rostan, G. [43] | 2003 | 4/49 | 8.2 | 16/29 | 55.2 | 15/29 | 51.7 |
| Hou, P. [55] | 2008 | 22/172 | 12.8 | - | - | 4/50 | 8.0 |
| Basolo, F. [56] | 2000 | 5/36 | 13.9 | 8/44 | 18.2 | 3/8 | 37.5 |
| Total | 73/492 | 14.8 | 48/173 | 27.7 | 30/169 | 17.7 |
Some studies have reported the association between RAS mutations and more aggressive behavior of thyroid cancer and a higher frequency of distant metastases. Five (33%) of 15 patients who died of PTC exhibited an RAS mutation, whereas this mutation was present in only eight (10.5%) of the 76 patients still alive after a 35 year follow-up [57]. In a 2012 study focused on 58 FTC, Fukahori et al. [58] found RAS mutations associated with distant metastases and poor overall survival. More recently, a study of 56 FTC, half of which with distant metastases, found an independent relationship between mutations at codon 61 of NRAS and distant metastases [59]. However, a limit of all mentioned studies is the low numerosity of the samples.
A large retrospective study including 1510 PTC followed for a mean of 33 months demonstrated less aggressive histological features, lower rates of extrathyroidal extension, and lower stage for RAS positive tumors, as compared to those with BRAF V600E or RET/PTC [60]. However, TERT promoter mutations, which were recently demonstrated to be of strong prognostic significance [61][62][63][64], were not included in the analysis, and this represents a crucial limitation. The analysis of the impact of RAS mutation on clinicopathological outcome in 162 BRAF mutation-negative PTC patients unveiled that RAS mutation alone had no adverse effects [65][66]. The patients harboring an RAS mutation alone did not display a more frequent tumor recurrence or higher mortality rate when compared with patients with no RAS, BRAF or TERT promoter mutation, suggesting that RAS mutation alone is not associated with aggressive behavior. Notably, the coexistence of RAS mutations with TERT promoter mutations was strongly associated with late disease stages, distant metastasis, and recurrence, as compared with the group of PTC negative for either mutation. These data, in addition with the finding that RAS mutations is more frequent in NIFTP than in other differentiated thyroid carcinomas, support the notion that RAS mutation alone is not a risk factor for a worse prognosis, whereas its coexistence with additional mutations is associated with a poorer outcome.
References
- Lemoine, N.R.; Mayall, E.S.; Wyllie, F.S.; Williams, E.D.; Goyns, M.; Stringer, B.; Wynford-Thomas, D. High frequency of ras oncogene activation in all stages of human thyroid tumorigenesis. Oncogene 1989, 4, 159–164.
- Namba, H.; Rubin, S.A.; Fagin, J.A. Point mutations of ras oncogenes are an early event in thyroid tumorigenesis. Mol. Endocrinol. 1990, 4, 1474–1479.
- Shi, Y.F.; Zou, M.J.; Schmidt, H.; Juhasz, F.; Stensky, V.; Robb, D.; Farid, N.R. High rates of ras codon 61 mutation in thyroid tumors in an iodide-deficient area. Cancer Res. 1991, 51, 2690–2693.
- Yoshimoto, K.; Iwahana, H.; Fukuda, A.; Sano, T.; Katsuragi, K.; Kinoshita, M.; Saito, S.; Itakura, M. Ras mutations in endocrine tumors: Mutation detection by polymerase chain reaction-single strand conformation polymorphism. Jpn. J. Cancer Res. 1992, 83, 1057–1062.
- Manenti, G.; Pilotti, S.; Re, F.C.; Della Porta, G.; Pierotti, M.A. Selective activation of ras oncogenes in follicular and undifferentiated thyroid carcinomas. Eur. J. Cancer 1994, 30A, 987–993.
- Ezzat, S.; Zheng, L.; Kolenda, J.; Safarian, A.; Freeman, J.L.; Asa, S.L. Prevalence of activating ras mutations in morphologically characterized thyroid nodules. Thyroid 1996, 6, 409–416.
- Naito, H.; Pairojkul, C.; Kitahori, Y.; Yane, K.; Miyahara, H.; Konishi, N.; Matsunaga, T.; Hiasa, Y. Different ras gene mutational frequencies in thyroid papillary carcinomas in Japan and Thailand. Cancer Lett. 1998, 131, 171–175.
- Bouras, M.; Bertholon, J.; Dutrieux-Berger, N.; Parvaz, P.; Paulin, C.; Revol, A. Variability of Ha-ras (codon 12) proto-oncogene mutations in diverse thyroid cancers. Eur. J. Endocrinol. 1998, 139, 209–216.
- Sugg, S.L.; Ezzat, S.; Zheng, L.; Freeman, J.L.; Rosen, I.B.; Asa, S.L. Oncogene profile of papillary thyroid carcinoma. Surgery 1999, 125, 46–52.
- Esapa, C.T.; Johnson, S.J.; Kendall-Taylor, P.; Lennard, T.W.; Harris, P.E. Prevalence of Ras mutations in thyroid neoplasia. Clin. Endocrinol. 1999, 50, 529–535.
- Motoi, N.; Sakamoto, A.; Yamochi, T.; Horiuchi, H.; Motoi, T.; Machinami, R. Role of ras mutation in the progression of thyroid carcinoma of follicular epithelial origin. Pathol. Res. Pr. 2000, 196, 1–7.
- Nikiforova, M.N.; Lynch, R.A.; Biddinger, P.W.; Alexander, E.K.; Dorn, G.W.; Tallini, G.; Kroll, T.G.; Nikiforov, Y.E. RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: Evidence for distinct molecular pathways in thyroid follicular carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 2318–2326.
- Vasko, V.; Ferrand, M.; Di Cristofaro, J.; Carayon, P.; Henry, J.F.; de Micco, C. Specific pattern of RAS oncogene mutations in follicular thyroid tumors. J. Clin. Endocrinol. Metab. 2003, 88, 2745–2752.
- Soares, P.; Trovisco, V.; Rocha, A.S.; Lima, J.; Castro, P.; Preto, A.; Maximo, V.; Botelho, T.; Seruca, R.; Sobrinho-Simoes, M. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene 2003, 22, 4578–4580.
- Garcia-Rostan, G.; Zhao, H.; Camp, R.L.; Pollan, M.; Herrero, A.; Pardo, J.; Wu, R.; Carcangiu, M.L.; Costa, J.; Tallini, G. ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J. Clin. Oncol. 2003, 21, 3226–3235.
- Zhu, Z.; Gandhi, M.; Nikiforova, M.N.; Fischer, A.H.; Nikiforov, Y.E. Molecular profile and clinical-pathologic features of the follicular variant of papillary thyroid carcinoma. An unusually high prevalence of ras mutations. Am. J. Clin. Pathol. 2003, 120, 71–77.
- Liu, R.T.; Hou, C.Y.; You, H.L.; Huang, C.C.; Hock, L.; Chou, F.F.; Wang, P.W.; Cheng, J.T. Selective occurrence of ras mutations in benign and malignant thyroid follicular neoplasms in Taiwan. Thyroid 2004, 14, 616–621.
- Hara, H.; Fulton, N.; Yashiro, T.; Ito, K.; DeGroot, L.J.; Kaplan, E.L. N-ras mutation: An independent prognostic factor for aggressiveness of papillary thyroid carcinoma. Surgery 1994, 116, 1010–1016.
- Park, J.Y.; Kim, W.Y.; Hwang, T.S.; Lee, S.S.; Kim, H.; Han, H.S.; Lim, S.D.; Kim, W.S.; Yoo, Y.B.; Park, K.S. BRAF and RAS mutations in follicular variants of papillary thyroid carcinoma. Endocr. Pathol. 2013, 24, 69–76.
- Schulten, H.J.; Salama, S.; Al-Ahmadi, A.; Al-Mansouri, Z.; Mirza, Z.; Al-Ghamdi, K.; Al-Hamour, O.A.; Huwait, E.; Gari, M.; Al-Qahtani, M.H.; et al. Comprehensive survey of HRAS, KRAS, and NRAS mutations in proliferative thyroid lesions from an ethnically diverse population. Anticancer Res. 2013, 33, 4779–4784.
- Giordano, T.J.; Beaudenon-Huibregtse, S.; Shinde, R.; Langfield, L.; Vinco, M.; Laosinchai-Wolf, W.; Labourier, E. Molecular testing for oncogenic gene mutations in thyroid lesions: A case-control validation study in 413 postsurgical specimens. Hum. Pathol. 2014, 45, 1339–1347.
- McFadden, D.G.; Dias-Santagata, D.; Sadow, P.M.; Lynch, K.D.; Lubitz, C.; Donovan, S.E.; Zheng, Z.; Le, L.; Iafrate, A.J.; Daniels, G.H. Identification of oncogenic mutations and gene fusions in the follicular variant of papillary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2014, 99, E2457–E2462.
- Jeong, S.H.; Hong, H.S.; Lee, E.H.; Kwak, J.J.; Lee, J.Y. Analysis of RAS mutation in thyroid nodular hyperplasia and follicular neoplasm in a Korean population. Endocrinol. Diabetes Metab. 2018, 1, e00040.
- Duan, H.; Liu, X.; Ren, X.; Zhang, H.; Wu, H.; Liang, Z. Mutation profiles of follicular thyroid tumors by targeted sequencing. Diagn. Pathol. 2019, 14, 39.
- Suarez, H.G.; Du Villard, J.A.; Caillou, B.; Schlumberger, M.; Tubiana, M.; Parmentier, C.; Monier, R. Detection of activated ras oncogenes in human thyroid carcinomas. Oncogene 1988, 2, 403–406.
- Du Villard, J.A.; Schlumberger, M.; Wicker, R.; Caillou, B.; Rochefort, P.; Feunteun, J.; Monier, R.; Parmentier, C.; Suarez, H.G. Role of ras and gsp oncogenes in human epithelial thyroid tumorigenesis. J. Endocrinol. Investig. 1995, 18, 124–126.
- Ricarte-Filho, J.C.; Ryder, M.; Chitale, D.A.; Rivera, M.; Heguy, A.; Ladanyi, M.; Janakiraman, M.; Solit, D.; Knauf, J.A.; Tuttle, R.M.; et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009, 69, 4885–4893.
- Liu, Z.; Hou, P.; Ji, M.; Guan, H.; Studeman, K.; Jensen, K.; Vasko, V.; El-Naggar, A.K.; Xing, M. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J. Clin. Endocrinol. Metab. 2008, 93, 3106–3116.
- Santarpia, L.; El-Naggar, A.K.; Cote, G.J.; Myers, J.N.; Sherman, S.I. Phosphatidylinositol 3-kinase/akt and ras/raf-mitogen-activated protein kinase pathway mutations in anaplastic thyroid cancer. J. Clin. Endocrinol. Metab. 2008, 93, 278–284.
- Hou, P.; Ji, M.; Xing, M. Association of PTEN gene methylation with genetic alterations in the phosphatidylinositol 3-kinase/AKT signaling pathway in thyroid tumors. Cancer 2008, 113, 2440–2447.
- Basolo, F.; Pisaturo, F.; Pollina, L.E.; Fontanini, G.; Elisei, R.; Molinaro, E.; Iacconi, P.; Miccoli, P.; Pacini, F. N-ras mutation in poorly differentiated thyroid carcinomas: Correlation with bone metastases and inverse correlation to thyroglobulin expression. Thyroid 2000, 10, 19–23.
- Bouras, M.; Bertholon, J.; Dutrieux-Berger, N.; Parvaz, P.; Paulin, C.; Revol, A. Variability of Ha-ras (codon 12) proto-oncogene mutations in diverse thyroid cancers. Eur. J. Endocrinol. 1998, 139, 209–216.
- Liu, R.T.; Hou, C.Y.; You, H.L.; Huang, C.C.; Hock, L.; Chou, F.F.; Wang, P.W.; Cheng, J.T. Selective occurrence of ras mutations in benign and malignant thyroid follicular neoplasms in Taiwan. Thyroid 2004, 14, 616–621.
- Manenti, G.; Pilotti, S.; Re, F.C.; Della Porta, G.; Pierotti, M.A. Selective activation of ras oncogenes in follicular and undifferentiated thyroid carcinomas. Eur. J. Cancer 1994, 30A, 987–993.
- Giordano, T.J.; Beaudenon-Huibregtse, S.; Shinde, R.; Langfield, L.; Vinco, M.; Laosinchai-Wolf, W.; Labourier, E. Molecular testing for oncogenic gene mutations in thyroid lesions: A case-control validation study in 413 postsurgical specimens. Hum. Pathol. 2014, 45, 1339–1347.
- Park, J.Y.; Kim, W.Y.; Hwang, T.S.; Lee, S.S.; Kim, H.; Han, H.S.; Lim, S.D.; Kim, W.S.; Yoo, Y.B.; Park, K.S. BRAF and RAS mutations in follicular variants of papillary thyroid carcinoma. Endocr. Pathol. 2013, 24, 69–76.
- McFadden, D.G.; Dias-Santagata, D.; Sadow, P.M.; Lynch, K.D.; Lubitz, C.; Donovan, S.E.; Zheng, Z.; Le, L.; Iafrate, A.J.; Daniels, G.H. Identification of oncogenic mutations and gene fusions in the follicular variant of papillary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2014, 99, E2457–E2462.
- Schulten, H.J.; Salama, S.; Al-Ahmadi, A.; Al-Mansouri, Z.; Mirza, Z.; Al-Ghamdi, K.; Al-Hamour, O.A.; Huwait, E.; Gari, M.; Al-Qahtani, M.H.; et al. Comprehensive survey of HRAS, KRAS, and NRAS mutations in proliferative thyroid lesions from an ethnically diverse population. Anticancer Res. 2013, 33, 4779–4784.
- Jeong, S.H.; Hong, H.S.; Lee, E.H.; Kwak, J.J.; Lee, J.Y. Analysis of RAS mutation in thyroid nodular hyperplasia and follicular neoplasm in a Korean population. Endocrinol. Diabetes Metab. 2018, 1, e00040.
- Schlumberger, M.J. Papillary and follicular thyroid carcinoma. N. Engl. J. Med. 1998, 338, 297–306.
- Zhu, Z.; Gandhi, M.; Nikiforova, M.N.; Fischer, A.H.; Nikiforov, Y.E. Molecular profile and clinical-pathologic features of the follicular variant of papillary thyroid carcinoma. An unusually high prevalence of ras mutations. Am. J. Clin. Pathol. 2003, 120, 71–77.
- Adeniran, A.J.; Zhu, Z.; Gandhi, M.; Steward, D.L.; Fidler, J.P.; Giordano, T.J.; Biddinger, P.W.; Nikiforov, Y.E. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am. J. Surg. Pathol. 2006, 30, 216–222.
- Gupta, S.; Ajise, O.; Dultz, L.; Wang, B.; Nonaka, D.; Ogilvie, J.; Heller, K.S.; Patel, K.N. Follicular variant of papillary thyroid cancer: Encapsulated, nonencapsulated, and diffuse: Distinct biologic and clinical entities. Arch. Otolaryngol. Head Neck Surg. 2012, 138, 227–233.
- Zhao, L.; Dias-Santagata, D.; Sadow, P.M.; Faquin, W.C. Cytological, molecular, and clinical features of noninvasive follicular thyroid neoplasm with papillary-like nuclear features versus invasive forms of follicular variant of papillary thyroid carcinoma. Cancer Cytopathol. 2017, 125, 323–331.
- Alexander, E.K.; Hurwitz, S.; Heering, J.P.; Benson, C.B.; Frates, M.C.; Doubilet, P.M.; Cibas, E.S.; Larsen, P.R.; Marqusee, E. Natural history of benign solid and cystic thyroid nodules. Ann. Intern. Med. 2003, 138, 315–318.
- Kuma, K.; Matsuzuka, F.; Yokozawa, T.; Miyauchi, A.; Sugawara, M. Fate of untreated benign thyroid nodules: Results of long-term follow-up. World J. Surg. 1994, 18, 495–498, discussion 499.
- Durante, C.; Costante, G.; Lucisano, G.; Bruno, R.; Meringolo, D.; Paciaroni, A.; Puxeddu, E.; Torlontano, M.; Tumino, S.; Attard, M.; et al. The natural history of benign thyroid nodules. JAMA 2015, 313, 926–935.
- Guerra, A.; Di Crescenzo, V.; Garzi, A.; Cinelli, M.; Carlomagno, C.; Tonacchera, M.; Zeppa, P.; Vitale, M. Genetic mutations in the treatment of anaplastic thyroid cancer: A systematic review. BMC Surg. 2013, 13 (Suppl. 2), S44.
- Basolo, F.; Pisaturo, F.; Pollina, L.E.; Fontanini, G.; Elisei, R.; Molinaro, E.; Iacconi, P.; Miccoli, P.; Pacini, F. N-ras mutation in poorly differentiated thyroid carcinomas: Correlation with bone metastases and inverse correlation to thyroglobulin expression. Thyroid 2000, 10, 19–23.
- Garcia-Rostan, G.; Zhao, H.; Camp, R.L.; Pollan, M.; Herrero, A.; Pardo, J.; Wu, R.; Carcangiu, M.L.; Costa, J.; Tallini, G. ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J. Clin. Oncol. 2003, 21, 3226–3235.
- Hara, H.; Fulton, N.; Yashiro, T.; Ito, K.; DeGroot, L.J.; Kaplan, E.L. N-ras mutation: An independent prognostic factor for aggressiveness of papillary thyroid carcinoma. Surgery 1994, 116, 1010–1016.
- Fukahori, M.; Yoshida, A.; Hayashi, H.; Yoshihara, M.; Matsukuma, S.; Sakuma, Y.; Koizume, S.; Okamoto, N.; Kondo, T.; Masuda, M.; et al. The associations between RAS mutations and clinical characteristics in follicular thyroid tumors: New insights from a single center and a large patient cohort. Thyroid 2012, 22, 683–689.
- Jang, E.K.; Song, D.E.; Sim, S.Y.; Kwon, H.; Choi, Y.M.; Jeon, M.J.; Han, J.M.; Kim, W.G.; Kim, T.Y.; Shong, Y.K.; et al. NRAS codon 61 mutation is associated with distant metastasis in patients with follicular thyroid carcinoma. Thyroid 2014, 24, 1275–1281.
- Yip, L.; Nikiforova, M.N.; Yoo, J.Y.; McCoy, K.L.; Stang, M.T.; Armstrong, M.J.; Nicholson, K.J.; Ohori, N.P.; Coyne, C.; Hodak, S.P.; et al. Tumor genotype determines phenotype and disease-related outcomes in thyroid cancer: A study of 1510 patients. Ann. Surg. 2015, 262, 519–525.
- Yoo, S.K.; Song, Y.S.; Lee, E.K.; Hwang, J.; Kim, H.H.; Jung, G.; Kim, Y.A.; Kim, S.J.; Cho, S.W.; Won, J.K.; et al. Integrative analysis of genomic and transcriptomic characteristics associated with progression of aggressive thyroid cancer. Nat. Commun. 2019, 10, 2764.
- Song, Y.S.; Lim, J.A.; Choi, H.; Won, J.K.; Moon, J.H.; Cho, S.W.; Lee, K.E.; Park, Y.J.; Yi, K.H.; Park, D.J.; et al. Prognostic effects of TERT promoter mutations are enhanced by coexistence with BRAF or RAS mutations and strengthen the risk prediction by the ATA or TNM staging system in differentiated thyroid cancer patients. Cancer 2016, 122, 1370–1379.
- Liu, R.; Xing, M. TERT promoter mutations in thyroid cancer. Endocr. Relat. Cancer 2016, 23, R143–R155.
- Yin, D.T.; Yu, K.; Lu, R.Q.; Li, X.; Xu, J.; Lei, M.; Li, H.; Wang, Y.; Liu, Z. Clinicopathological significance of TERT promoter mutation in papillary thyroid carcinomas: A systematic review and meta-analysis. Clin. Endocrinol. 2016, 85, 299–305.
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690.
- Shen, X.; Liu, R.; Xing, M. A six-genotype genetic prognostic model for papillary thyroid cancer. Endocr. Relat. Cancer 2017, 24, 41–52.

