 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Donald E. Spratt | + 2318 word(s) | 2318 | 2021-08-24 08:26:51 | | | |
| 2 | Camila Xu | + 388 word(s) | 2706 | 2021-08-30 06:08:16 | | | | |
| 3 | Camila Xu | + 388 word(s) | 2706 | 2021-08-30 06:08:55 | | |
Video Upload Options
The ubiquitin-proteasome system (UPS) is a highly regulated mechanism for protein degradation that regulates many biological processes to maintain cellular homeostasis. A protein is targeted for degradation upon ubiquitylation, where the small 8.6 kDa protein, ubiquitin, is covalently attached to the target protein through an isopeptide bond. Ubiquitylation involves the sequential transfer of ubiquitin through a three-enzyme cascade—an ubiquitin-activating enzyme (E1), an ubiquitin-conjugating enzyme (E2), and an ubiquitin ligase (E3).
1. Introduction
Neurodegenerative disease (NDD) research initiated approximately four decades ago, but recently, the focus has shifted to understanding both the neurochemistry aspect in disease development and neuroprotection. Proper central nervous system (CNS) development plays an essential role in neuroprotection, as any malformation during this intricate process allows for an increased susceptibility to neurodegeneration. The foundation of the CNS occurs during embryogenesis, where approximately 40% of the developing genes primarily regulate CNS development [1]. This starts with the development of the neural plate, which continues to grow and fold onto itself until this groove forms into the neural tube [2]. This developmental precursor relies on specific signals to induce rudimentary CNS formation [2]. At the final stages of development, the CNS can be divided into two sections—the brain and the spinal cord—with both involved in receiving and processing sensory information to induce a biological response [3]. While both the brain and spinal cord are encapsulated within bone, the meninges and cerebrospinal fluid (CSF) act as physical barriers with biochemical protective factors to prevent degeneration [3]. In addition to the meninges and CSF, signaling proteins are essential to the regulation of neuroprotective pathways, simultaneously providing a system of checks and balances to protect the CNS from neurodegeneration. For example, autophagic regulating proteins and ubiquitin were found to have altered expression levels in the CSF of Alzheimer’s disease (AD) and Parkinson’s disease (PD) patients [4][5][6][7].
Consequential neuronal death due to varying biological irregularities lead to the onset of various NDDs such as amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), AD, PD, and Huntington’s disease (HD) [8]. Both AD and PD pathology are caused by the accumulation of protein aggregates resulting from the dysfunction in varying cellular pathways. In AD pathology, it is suggested that an accumulation of autophagic vacuoles are present in the cortex due to an inability to perform or an inhibition of cholinergic neuron mitophagy [4][9][10]. In contrast, PD involves the hallmark histopathological Lewy body detection in mitochondrial-based proteins associated with degraded dopaminergic neurons [4][11][12]. While it is unclear if misfolded proteins contribute to disease development upon aggregation or are merely signs of proper biological processes that gather and eliminate harmful proteins, the etiology is still elusive ( Figure 1 ). It is clear, however, that disrupting neuronal autophagy or progressing presynaptic neuron death results in NDD pathogenesis [8][9][10][11][12][13].
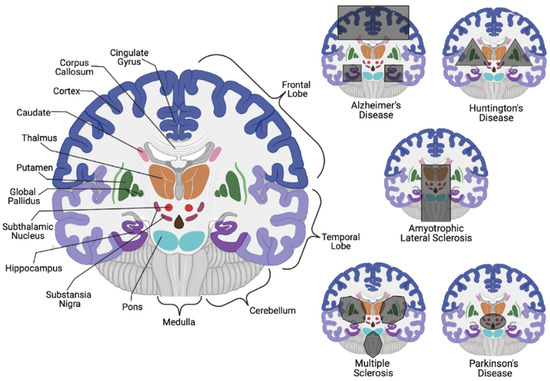
While correlations exist for both AD and PD on a genetic and hereditary level, it is still inconclusive whether these diseases are inherited or idiopathic. On the other hand, HD is an autosomal dominant NDD that impacts motor skills and cognition through neuronal loss in the striatum, with an average 15-year life expectancy post-diagnosis [20]. Striatum neuronal degeneration is believed to be triggered by CAG trinucleotide tandem repeats that result in extended polyglutamine sequences in the translated huntingtin protein (Htt). While 6 to 10 polyglutamine sequences in eukaryotic proteins have been identified to facilitate protein-protein interactions (PPIs) through the coiled-coil conjunction within two proteins [21], the CAG trinucleotide repeats within Htt are typically 10–35 units in length; the abnormal gain of function HD-derived mutations allow for upwards of 40–100 repeats that alter Htt localization and accumulation [22][23]. The function of Htt remains experimentally unclear, but its cerebral and basal ganglia localization in the brain suggests its involvement in the regulation of movement coordination.
During embryogenesis, Htt levels are evenly dispersed throughout progenitor cells that eventually differentiate into striatum-based cortical neurons, whereas in HD fetuses, the Htt protein congregates in the apical end-feet of progenitor cells [24]. This suggests that Htt may play a role in proper CNS development by ensuring proper motor skill development and coordination through the facilitation of progenitor cell polarity and differentiation during neurogenesis. The striatum, being the input model for the basal ganglia, emits signals to dictate learning through action selection and behavior reinforcement [13][25][26]. Classified as a basal ganglia disease, HD phenotypes are typically subtle, with a slow progression, until the patient is unable to move and speak [20]. It can be speculated excess CAG tandem repeats inhibit mutant Htt (mHtt) aggregate clearance and impair essential neurodevelopment-based PPIs due to conformational changes; this is similar to other neurological diseases that lead to aggregate buildup and mitochondrial defects [27]. As observed in HD mice models, developmental pathways that regulate synaptic homeostasis are impaired [28]. Htt has been identified to interact with numerous developmental proteins that regulate 14-3-3 signaling, microtubule-based transport, and proteostasis [29]. A structural analysis of mHtt in any of these essential developmental proteins can determine if the excess CAG repeats truly alter these PPIs, or even whether downstream activation of other pathways is also impaired.
2. Neuroprotection through Redox Chemistry
From development to the senescent stages of the CNS, metabolically driven processes lead towards excess electrophilic byproducts; thus, alleviating this accumulation is essential for biological homeostasis. Metabolically driven processes in the brain required for proper function account for approximately 20% of the overall energy consumption in the body [30], and these reactions require a properly regulated mitochondrial electron transfer to produce sufficient amounts of ATP. As electrons are passed through the different complexes in the electron transport chain (ETC), both H+ and H2O are produced from O2 reduction by cytochrome c oxidase [31]. The inability to properly reduce O2 can cause the production of damaging reactive oxygen species (ROS).
Redox homeostasis helps to the maintain the balance between the production of antioxidants, ROS, and some reactive nitrogen species (RNS) [32]. Many ROS, which includes hydrogen peroxide (H2O2), hydroxyl radical (•OH), hydroperoxyl radical (HO2•), peroxyl radical (ROO•), superoxide (O2•− ), and singlet oxygen (1O2), are naturally occurring byproducts of mitochondrial metabolism ( Figure 2 ) [33].
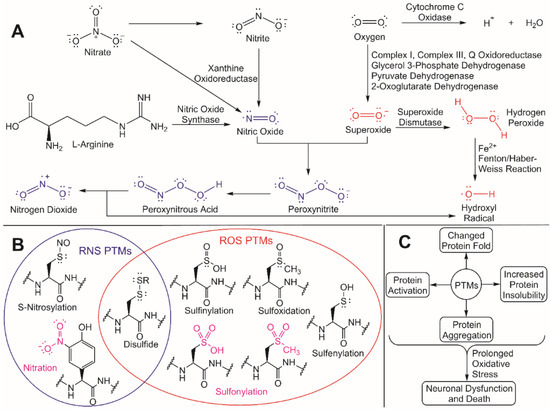
Despite this regulatory role, unregulated ROS accumulation can trigger oxidative stress and an increased nucleophilic attack of exposed residues in proteins that are susceptible to ROS/RNS modifications [33][41]. These ROS/RNS-induced PTMs can cause different changes in a protein’s fold and/or function, including increased insolubility and subsequent aggregation due to the exposure of hydrophobic residues in the modified protein [42]. Cysteine residues are particularly susceptible to ROS/RNS modification due to the inherent hyperreactivity of the R-group thiol [43]. If oxidative stress is prolonged and aggregated proteins continue to accumulate, neuronal dysfunction and death can occur, which is a hallmark in numerous NDD. To better understand how to protect against neurodegeneration, it is critical to understand both the molecular effects that ROS has on cellular processes and the underlying mechanisms for how redox homeostasis regulators are activated. These unregulated ROS/RNS PTMs can be mitigated through ubiquitylation by activating the redox homeostasis response, thus leading to the degradation and eventual clearance of protein aggregates.
Protein modifications that are enhanced by RNS accumulation have detrimental impacts on neuroprotection. For example, RNS accumulation after a traumatic brain injury (TBI) is speculated to induce the S-nitrosylation of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). With glyceraldehyde 3-phosphate dehydrogenase (GAPDH), RNS accumulation following traumatic brain injury (TBI) is speculated to influence S-nitrosylation. Through GADPH S-nitrosylation, Sirtuin1 deacetylation is prevented, which then activates p300/CBP acetyltransferase [44]. This leads to TBI-induced tau acetylation at Lys274 and Lys281. The modification of tau at these lysine residues is also observed in AD [45], further suggesting the essential role of regulating RNS-induced PTMs to alleviate both aging and non-aging neurodegeneration. It would therefore be imperative to assess the potential of targeting S-nitrosylated GAPDH for proteolysis as a therapeutic approach for alleviating tauopathy memory loss, AD, or other tau-related NDDs.
3. Interplay between the Ubiquitin-Proteasome System and ROS Production
The ubiquitin-proteasome system (UPS) is a highly regulated mechanism for protein degradation that regulates many biological processes to maintain cellular homeostasis [46]. A protein is targeted for degradation upon ubiquitylation, where the small 8.6 kDa protein, ubiquitin, is covalently attached to the target protein through an isopeptide bond [47]. Ubiquitylation involves the sequential transfer of ubiquitin through a three-enzyme cascade—an ubiquitin-activating enzyme (E1), an ubiquitin-conjugating enzyme (E2), and an ubiquitin ligase (E3). The E2/E3 combination ultimately determines the specific site of attachment of ubiquitin on the target protein and the lysine linkage between ubiquitin molecules within the polyubiquitin chain that is built [47][48][49][50]. The process is initiated when ubiquitin and E1 undergo an ATP-dependent reaction in which a thioester bond is formed between the C-terminus of ubiquitin and the catalytic cysteine on the E1 (E1~ubiquitin) [46]. The activated ubiquitin is then transferred through a transthiolation reaction to a cysteine residue on an E2 enzyme (E2~ubiquitin) [50]. An E3 ligase then facilitates or directly catalyzes the transfer of ubiquitin from the E2~ubiquitin complex to a lysine residue of the substrate to form a stable isopeptide bond between the C-terminus of ubiquitin and the epsilon-amine of the lysine residue on the substrate [50][51]. The type of mechanism used to attach ubiquitin onto a target protein is dependent on the type of E3 ligase—Really Interesting New Gene (RING), Homologous to E6-AP Carboxyl Terminus (HECT), or RING-Between-RING (RBR) [46]. A RING E3 ubiquitin ligase acts as a scaffold to mediate the transfer of ubiquitin from the E2~ubiquitin complex directly to a lysine residue on the substrate [47]. In contrast, a HECT E3 ubiquitin ligase removes ubiquitin from E2~ubiquitin to form an E3~ubiquitin thioester intermediate, which then catalyzes the ubiquitin transfer on to the substrate [46]. A RBR E3 ubiquitin ligase recruits an E2~ubiquitin complex through its RING1 domain, like a RING E3 ligase, but instead of coordinating the transfer of ubiquitin from E2~ubiquitin directly to a substrate, the ubiquitin is transferred to a conserved catalytic cysteine in the RING2 domain (also known as “required for catalysis” (Rcat)) prior to discharge on to a substrate, analogous to the HECT E3 ubiquitin ligase mechanism [52].
Ubiquitylation plays an important role in maintaining cellular homeostasis by regulating the activities of critical ROS eliminating proteins through transcription activation. What makes this mechanism of Nrf2 regulation interesting is the requirement of the Cullin-3/Keap1/Rbx1 complex, further supporting the multifunctional roles these proteins play in neuroprotection beyond direct protein– protein interactions for Nrf2 signaling pathway activation or inhibition. Disrupting or even enhancing necessary large protein complexes can serve as a therapeutic approach towards NDD treatment; this, however, requires a full understanding, on a biophysical and biochemical level, of how these complexes form exactly and which residues of each protein are essential to solidify various bonds and interactions.
Nitric oxide (NO) accumulation also activates a cascading signal to induce apoptosis in the event that NO accumulation cannot be properly regulated. Brain-derived neurotrophic factor (BDNF) induces NO production and increases cAMP response element-binding protein (CREB) binding activity, a critical regulator of gene expression in dopaminergic neurons [53]. With CREB activation, GAPDH undergoes S-nitrosylation at Cys150, which abolishes GAPDH activity [54]. This irregular PTM leaves GAPDH susceptible to unstable absentia homolog 1 (SIAH1) binding, which in turn leads to SIAH1-dependent ubiquitylation of GAPDH to induce apoptosis [55][56]. SIAH1 can also activate dopamine release through the inhibition of synphilin-1, the primary neurotransmitter depleted in PD patients [55]. However, synphilin-1 is arguably also neuroprotective since it inhibits cytochrome c translocation, MPP+ based ROS production, and apoptosis in PD-impacted dopaminergic neurons [57]. Regardless of potential neuroprotective capabilities, elevated protein levels can disrupt cellular homeostasis and must be tightly regulated through ubiquitylation to control cell death.
Neuroprotection can also be fulfilled with fibrous or aggregate clearance through proteolysis, autophagic, or lysosomal pathways, with all variations of aggregate clearance heavily dependent on the proper and timely covalent attachment of ubiquitin to a target protein. A myriad of signaling pathways are activated through ubiquitylation, including the classic proteolysis of a specific substrate through the 26S proteasome [46]. Various mechanisms that drive this protective function are primarily facilitated through ubiquitylation to cause their colocalization to areas of protein aggregation within the cell, which is not their typical function. It can be speculated that various proteins involved in ubiquitylation may have adapted multifunctional capabilities in order to further provide neuroprotective properties. A prime example is TRAF6, which was found to colocalize in Lewy bodies to facilitate the cytoplasmic aggregation of both Parkinson disease protein 7 (DJ-1/PARK7) and α-synuclein caused by atypical ubiquitylation via K6-, K27-, and K29-ubiquitin linkages [58]. While ubiquitylation for aggregate accumulation are typically caused K63-ubiquitin linkages, both K6- and K29-linkages retain a relatively linear chain formation but a more compact fold, possibly to encourage aggregate clearance and potentially prevent their untimely deubiquitylation.
4. NDD Regulation by ROS and the UPS: Moving Forward
Ubiquitylation plays a critical role in neuroprotection, far beyond its proteolytic degradation targeting activity. This modification is essential in neurodevelopment, neuronal homeostasis mediation, and the prevention of aging-related neurodegeneration. As discussed, this form of a PTM not only targets proteins for degradation, but also utilizes ubiquitin attachment to trigger conformational changes, activates aggregate clearance pathways such as autophagy, maintains CNS glial cell function through its support of complex formations, and enhances protein nuclear trafficking for transcription activation. Various pathways also rely on the properly timed modifications of substrates to regulate the intracellular accumulation of reactive species in preventing neurodegeneration. However, reactive species are also imperative for cell differentiation, cell–cell signaling, the proper timing for the activation of such pathways, and maintaining proper neurotransmitter production. ROS and RNS levels need to be properly regulated since total removal of ROS and RNS would inhibit some of the essential cellular mechanisms that we have discussed in this review. Maintaining ROS levels is essential for homeostasis. Although the current research focus on NDD pathology remains centered on improper protein folding, these recent findings that show the intricate cross-communication between redox chemistry and ubiquitylation may reveal novel neuroprotective functions that could serve as new future NDD drug targets.
References
- Schmidt-Sidor, B.; Mierzewska, H.; Turzyniecka, M.; Kowalewska-Kantecka, B.; Wierzba-Bobrowicz, T.; Lechowicz, W. Neurodegenerative disease in infants with multiple congenital malformations—Report of two cases. Folia Neuropathol. 2004, 42, 221–226.
- Silbereis, J.C.; Pochareddy, S.; Zhu, Y.; Li, M.; Sestan, N. The Cellular and Molecular Landscapes of the Developing Human Central Nervous System. Neuron 2016, 89, 248–268.
- Thau, L.; Reddy, V.; Singh, P. Anatomy, Central Nervous System; StatPearls Publishing: Treasure Island, FL, USA, 2020.
- Sjodin, S.; Brinkmalm, G.; Ohrfelt, A.; Parnetti, L.; Paciotti, S.; Hansson, O.; Hardy, J.; Blennow, K.; Zetterberg, H.; Brinkmalm, A. Endo-lysosomal proteins and ubiquitin CSF concentrations in Alzheimer’s and Parkinson’s disease. Alzheimers Res. Ther. 2019, 11, 82.
- Arnold, J.; Dawson, S.; Fergusson, J.; Lowe, J.; Landon, M.; Mayer, R.J. Ubiquitin and its role in neurodegeneration. Prog. Brain Res. 1998, 117, 23–34.
- Huang, Q.; Figueiredo-Pereira, M.E. Ubiquitin/proteasome pathway impairment in neurodegeneration: Therapeutic implications. Apoptosis 2010, 15, 1292–1311.
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49.
- Heemels, M.T. Neurodegenerative diseases. Nature 2016, 539, 179.
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166.
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115.
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013, 47, 495–508.
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912.
- Roze, E.; Cahill, E.; Martin, E.; Bonnet, C.; Vanhoutte, P.; Betuing, S.; Caboche, J. Huntington’s Disease and Striatal Signaling. Front. Neuroanat. 2011, 5, 55.
- Lee, P.L.; Chou, K.H.; Chung, C.P.; Lai, T.H.; Zhou, J.H.; Wang, P.N.; Lin, C.P. Posterior Cingulate Cortex Network Predicts Alzheimer’s Disease Progression. Front. Aging Neurosci. 2020, 12, 608667.
- Reiner, A.; Dragatsis, I.; Dietrich, P. Genetics and neuropathology of Huntington’s disease. Int. Rev. Neurobiol. 2011, 98, 325–372.
- Pan, M.; Zheng, Q.; Yu, Y.; Ai, H.; Xie, Y.; Zeng, X.; Wang, C.; Liu, L.; Zhao, M. Seesaw conformations of Npl4 in the human p97 complex and the inhibitory mechanism of a disulfiram derivative. Nat. Commun. 2021, 12, 121.
- Minagar, A.; Barnett, M.H.; Benedict, R.H.; Pelletier, D.; Pirko, I.; Sahraian, M.A.; Frohman, E.; Zivadinov, R. The thalamus and multiple sclerosis: Modern views on pathologic, imaging, and clinical aspects. Neurology 2013, 80, 210–219.
- Liptak, Z.; Berger, A.M.; Sampat, M.P.; Charil, A.; Felsovalyi, O.; Healy, B.C.; Hildenbrand, P.; Khoury, S.J.; Weiner, H.L.; Bakshi, R.; et al. Medulla oblongata volume: A biomarker of spinal cord damage and disability in multiple sclerosis. AJNR Am. J. Neuroradiol. 2008, 29, 1465–1470.
- Prakash, K.G.; Bannur, B.M.; Chavan, M.D.; Saniya, K.; Sailesh, K.S.; Rajagopalan, A. Neuroanatomical changes in Parkinson’s disease in relation to cognition: An update. J. Adv. Pharm. Technol. Res. 2016, 7, 123–126.
- Vonsattel, J.P.; DiFiglia, M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384.
- Schaefer, M.H.; Wanker, E.E.; Andrade-Navarro, M.A. Evolution and function of CAG/polyglutamine repeats in protein-protein interaction networks. Nucleic Acids Res. 2012, 40, 4273–4287.
- Dayalu, P.; Albin, R.L. Huntington disease: Pathogenesis and treatment. Neurol. Clin. 2015, 33, 101–114.
- Frank, S. Treatment of Huntington’s disease. Neurotherapeutics 2014, 11, 153–160.
- Lempriere, S. Huntington disease alters early neurodevelopment. Nat. Rev. Neurol. 2020, 16, 459.
- Zheng, P.; Kozloski, J. Striatal Network Models of Huntington’s Disease Dysfunction Phenotypes. Front. Comput. Neurosci. 2017, 11, 70.
- Graybiel, A.M. The basal ganglia and chunking of action repertoires. Neurobiol. Learn. Mem. 1998, 70, 119–136.
- La Spada, A.R.; Paulson, H.L.; Fischbeck, K.H. Trinucleotide repeat expansion in neurological disease. Ann. Neurol. 1994, 36, 814–822.
- Consortium, H.D.I. Developmental alterations in Huntington’s disease neural cells and pharmacological rescue in cells and mice. Nat. Neurosci. 2017, 20, 648–660.
- Shirasaki, D.I.; Greiner, E.R.; Al-Ramahi, I.; Gray, M.; Boontheung, P.; Geschwind, D.H.; Botas, J.; Coppola, G.; Horvath, S.; Loo, J.A.; et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron 2012, 75, 41–57.
- Harris, J.J.; Attwell, D. The energetics of CNS white matter. J. Neurosci. 2012, 32, 356–371.
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261.
- Dasuri, K.; Zhang, L.; Keller, J.N. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic. Biol. Med. 2013, 62, 170–185.
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Opportunities. Antioxid. Redox Signal. 2019, 30, 1450–1499.
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804.
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049.
- Weitzberg, E.; Hezel, M.; Lundberg, J.O. Nitrate-nitrite-nitric oxide pathway: Implications for anesthesiology and intensive care. Anesthesiology 2010, 113, 1460–1475.
- Petushkova, A.I.; Zamyatnin, A.A., Jr. Redox-Mediated Post-Translational Modifications of Proteolytic Enzymes and Their Role in Protease Functioning. Biomolecules 2020, 10, 650.
- Goyal, M.M.; Basak, A. Hydroxyl radical generation theory: A possible explanation of unexplained actions of mammalian catalase. Int. J. Biochem. Mol. Biol. 2012, 3, 282–289.
- Chen, S.X.; Schopfer, P. Hydroxyl-radical production in physiological reactions. A novel function of peroxidase. Eur. J. Biochem. 1999, 260, 726–735.
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624.
- Ferrer-Sueta, G.; Manta, B.; Botti, H.; Radi, R.; Trujillo, M.; Denicola, A. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem. Res. Toxicol. 2011, 24, 434–450.
- Santos, A.L.; Lindner, A.B. Protein Posttranslational Modifications: Roles in Aging and Age-Related Disease. Oxid. Med. Cell. Longev. 2017, 2017, 5716409.
- Duan, J.; Gaffrey, M.J.; Qian, W.J. Quantitative proteomic characterization of redox-dependent post-translational modifications on protein cysteines. Mol. Biosyst. 2017, 13, 816–829.
- Shin, M.K.; Vazquez-Rosa, E.; Koh, Y.; Dhar, M.; Chaubey, K.; Cintron-Perez, C.J.; Barker, S.; Miller, E.; Franke, K.; Noterman, M.F.; et al. Reducing acetylated tau is neuroprotective in brain injury. Cell 2021, 184, 2715–2732.
- Tracy, T.E.; Sohn, P.D.; Minami, S.S.; Wang, C.; Min, S.W.; Li, Y.; Zhou, Y.; Le, D.; Lo, I.; Ponnusamy, R.; et al. Acetylated Tau Obstructs KIBRA-Mediated Signaling in Synaptic Plasticity and Promotes Tauopathy-Related Memory Loss. Neuron 2016, 90, 245–260.
- Wang, Y.; Argiles-Castillo, D.; Kane, E.I.; Zhou, A.; Spratt, D.E. Correction: HECT E3 ubiquitin ligases—Emerging insights into their biological roles and disease relevance. J. Cell Sci. 2020, 133, jcs258087.
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta 2014, 1843, 47–60.
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880.
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229.
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422.
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479.
- Spratt, D.E.; Walden, H.; Shaw, G.S. RBR E3 ubiquitin ligases: New structures, new insights, new questions. Biochem. J. 2014, 458, 421–437.
- Wang, H.; Xu, J.; Lazarovici, P.; Quirion, R.; Zheng, W. cAMP Response Element-Binding Protein (CREB): A Possible Signaling Molecule Link in the Pathophysiology of Schizophrenia. Front. Mol. Neurosci. 2018, 11, 255.
- Hara, M.R.; Agrawal, N.; Kim, S.F.; Cascio, M.B.; Fujimuro, M.; Ozeki, Y.; Takahashi, M.; Cheah, J.H.; Tankou, S.K.; Hester, L.D.; et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 2005, 7, 665–674.
- Nagano, Y.; Yamashita, H.; Takahashi, T.; Kishida, S.; Nakamura, T.; Iseki, E.; Hattori, N.; Mizuno, Y.; Kikuchi, A.; Matsumoto, M. Siah-1 facilitates ubiquitination and degradation of synphilin-1. J. Biol. Chem. 2003, 278, 51504–51514.
- Okamoto, S.; Lipton, S.A. S-Nitrosylation in neurogenesis and neuronal development. Biochim. Biophys. Acta 2015, 1850, 1588–1593.
- Shishido, T.; Nagano, Y.; Araki, M.; Kurashige, T.; Obayashi, H.; Nakamura, T.; Takahashi, T.; Matsumoto, M.; Maruyama, H. Synphilin-1 has neuroprotective effects on MPP(+)-induced Parkinson’s disease model cells by inhibiting ROS production and apoptosis. Neurosci. Lett. 2019, 690, 145–150.
- Zucchelli, S.; Codrich, M.; Marcuzzi, F.; Pinto, M.; Vilotti, S.; Biagioli, M.; Ferrer, I.; Gustincich, S. TRAF6 promotes atypical ubiquitination of mutant DJ-1 and alpha-synuclein and is localized to Lewy bodies in sporadic Parkinson’s disease brains. Hum. Mol. Genet. 2010, 19, 3759–3770.

