 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | John Mackrill | + 1925 word(s) | 1925 | 2021-08-16 13:37:10 |
Video Upload Options
Oxysterols are cholesterol oxidation products, which can be absorbed from the diet, or generated by auto-oxidation or by enzymatic mechanisms. Oxysterols result from oxidation of cholesterol on the sterol rings, the side chain, or both. This generates a diverse range of oxysterol congeners that have distinct biophysical properties.
1. Introduction
In addition to being reaction intermediates in the synthesis of bile-acids and steroid hormones, many oxysterols are biologically active signalling molecules, regulating diverse cellular processes. Consequently, the dysregulation of oxysterol production and action is associated with a range of diseases, including cancers [1], atherosclerosis [2], age-related macular degeneration, neurodegeneration and osteoporosis [3]. The focus of the current review are the roles of oxysterols in immune system physiology and pathology [4][5][6].
A key class of intracellular receptors for oxysterols exert their cellular effects through changes in the transcription of target genes [7]. Archetypal nuclear receptors for oxysterols are the liver X receptors (LXRs), which exist as heterodimers with retinoid X receptors (RXRs). Of the two human LXRs, LXRα (nuclear receptor subfamily 1, group H, member 3 (NR1H3)) is abundant in macrophages, liver, intestine, adipose tissue, lung, kidney and adrenal gland, whereas LXRβ (NR1H2) is ubiquitous [8]. Binding of certain side-chain oxysterols (22(R)-HC, 24(S)-HC, 25-HC, or 27-HC) elicits conformational changes that promote dissociation of LXR-RXR complexes from transcriptional co-repressors, and stimulates LXR-RXR interaction with the sterol response elements (SREs) of target genes. These mechanisms increase the transcription of genes involved in cholesterol clearance (ATP-binding cassette A1 and G1, CYP7A1 , apolipoprotein E) and those which regulate multiple components of the immune system [4].
There are three widely expressed members of the SRE-binding protein family (SREBP1a, SREBP1b and SREBP2), which are transcription factors tethered to the ER via a transmembrane-spanning domain. In the ER, SREBPs interact with SREBP cleavage-activating proteins (SCAPs). When the levels of cholesterol and oxysterols are low, SCAP transports SREBPs to the Golgi apparatus where it is cleaved by two resident proteases, releasing an N-terminal domain. This domain translocates to the nucleus, where it binds to SREs in the promoters of target genes [9]. These target genes include those involved in fatty acid metabolism, cholesterol transport and immune responses [6]. Cholesterol, or certain oxysterols (most notably 25-HC), bind to the ER-resident proteins insulin-induced gene-1 (INSIG-1) or INSIG-2, promoting interaction with SCAP [10], preventing its translocation to the Golgi and activation of SREBP. Consequently, cholesterol and some oxysterols are negative modulators of SREBP-dependent transcription.
Despite the potential for therapeutic exploitation of nuclear receptor-mediated oxysterol signalling in the regulation of immune cell biology, this does have two key limitations. Firstly, transcriptional mechanisms are slow, with a lag of tens of minutes to hours between the initial stimulus (change in the levels of a particular oxysterol) and the cellular response. This may be unsuitable for the control of cellular processes that can occur over much shorter timescales, such as motility, endocytosis and secretion. Secondly, in terms of the development of oxysterol-based immunomodulatory drugs, LXR agonists act on the liver to increase the expression of SREBP1c, which promotes the clinically unfavourable condition of hypertriglyceridemia [6][11][12]. Targeting the main focus of the current review, cell-surface receptors for oxysterols might provide effective solutions to these limitations. These targets can be divided into two classes: G protein-coupled receptors (GPRs) and ion channels.
2. G Protein-Coupled Receptors
G protein-coupled receptors (GPCRs) are predominantly located in the cell-surface membrane. They have a characteristic 7-transmembrane (7TM) topology and regulate the activities of second-messenger-producing enzymes. Figure 1 summarizes some of the properties of the three types of GPCR that are present in the mammalian immune system and which interact with oxysterols.
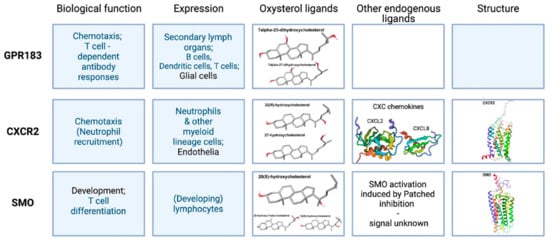
Comparing different structures resolved in recent years, some major differences can be seen in the tilt of the CRD region. This might be due to different activation states [20][21]. Deshpande et al. [22] solved a full receptor structure, stabilized in the active state by nanobody binding, with cholesterol bound in a deep pocket inside the 7TM region (PDB:6O3C). This binding produces the activating changes for GPCRs, independent of Hh, especially the marked shift in TM6. The binding pocket opens up with the receptor in a more active-like state; for example, with agonists bound higher up in the receptor, or by stimulation from the CRD site.
GABA is the main inhibitory neurotransmitter in the vertebrate central nervous system. It exerts biological effects by interacting with ionotropic GABAARs (see Section 3.1.3), which contain an intrinsic chloride channel, and with metabotropic GABABRs, which are GPCRs. There is limited evidence supporting a modulatory role for oxysterols in GABABR signalling. In brain slices from the rat lateral septum, the superfusion of 25-HC selectively reduced GABABR- but not GABAAR-dependent inhibitory post-synaptic potentials [23]. Microglia are known to possess both GABABR subtypes. The activation of these receptors inhibits microglial IL-6 and IL-12p40 secretion in response to LPS [24]. The GABABR-selective antagonist, baclofen, is reported to inhibit DC cell activation and their priming of Th17+ T cells [25].
3. Ion Channels
Gating of ion channels permits rapid changes in intracellular ion concentrations in response to diverse stimuli, leading to alterations in membrane potential, the second messenger Ca 2+ , cell volume, cell-death, gene expression, secretion, endocytosis, or motility. The gating of a limited range of ion channels is known to be modulated by oxysterols, often in a congener- and channel subtype-selective channel fashion. In addition to cell-surface channel proteins, IP 3R Ca 2+ release channels are located predominantly on the ER and are also modulated by oxysterols. This includes the stimulation of the proteolytic degradation of IP 3Rs [26], and the assembly of IP 3R signalling complexes by [27], and direct interactions with, ORP4L [28]. The cell-surface ion channels that are modulated by oxysterols can be divided into three functional categories: those gated by ligands; those gated by changes in membrane potential (voltage-gated); and those gated by multiple stimuli (multi-modal gating). The presence and roles of these channels in the immune system are largely unexplored. These channels have considerable potential as targets for the development of new therapies to combat immune disorders, including autoimmune diseases [4][5].
Negative modulation of NMDARs by some oxysterol congeners might provide an explanation for an absence of cytotoxicity in immune cells, in the face of high concentrations of excitatory amino acids encountered in the blood. NMDARs are present in some immune cells, as reviewed by Boldyrev in 2005 [29]. Using fluorescence-activated cytometry, GluN1 was detected in human peripheral lymphocytes, with the activation of NMDARs in these cells being associated with Ca 2+ influx, ROS production and cell death. In NK cells derived from this population, NMDA had no effect on IFN-γ production, but suppressed that stimulated by IL-2 [30]. In mouse splenic B-lymphocytes, non-competitive NMDAR antagonists reduced proliferation, chemotaxis and immunoglobulin secretion, but enhanced IL-10 production. However, these antagonists block K + currents through voltage-gated K v 1.3 and K Ca 3.1 present in these and other immune cells (see Section 3.2.1.1 and Section 3.3.1 ) [31][32]. These observations highlight the need for caution when interpreting the effects of small molecule drugs on immune cells.
Voltage-gated Na + channels have a canonical role in underlying action potentials in excitable tissues, but regulate cell-volume, motility, secretion and death in other cell types. They are comprised of a channel-forming, voltage-sensing α subunit (Na v 1.1–1.5 encoded by SCN1A–SCN5A , Na v 1.6 encoded by SCN8A , Na v 1.7–1.8 encoded by SCN9A–SCN11A, and Na x encoded by SCN7A) in combination with accessory proteins, such as the β-sub-units (Na v β1–4 encoded by SCN1B–SCN4B ), which influence channel gating or trafficking [33]. Canonical antagonists of these channels include the puffer-fish toxin, tetrodotoxin (TTX), and the small molecule drug, amiloride. Since voltage-gated Na + channels are present in lymphocyte- and monocyte-derived immune cells, there is potential for small molecule modulators of these channels to be utilized as immunotherapeutic drugs. This is offset by potentially severe side-effects of such drugs on the nervous system [34].
The opening of a subset of voltage-gated K + channels is stimulated by increased cytoplasmic Ca 2+ concentrations. A well-characterized member of this family is called K Ca 1.1, or Slo-1, the large-conductance K + channel BK, or MaxiK and is encoded by the calcium-activated K + channel subunit, subfamily M, alpha-1 ( KCNMA1 ) gene. BK channels are formed from tetrameric assemblies of K Ca 1.1 protein and their electrophysiology, trafficking and interactions with ligands are modulated by associations with accessory proteins, most notably β and γ subunits [35]. A key component of K Ca 1.1 gating is a switch from positively charged amino acids (the “RKK ring”) binding to negatively charged glutamate residues in the protein when in a closed state, to binding oxygen atoms of membrane lipids when in an open state. This gating mechanism helps explain the modulation of BK channel gating by lipophilic modulators [36]. BK gating can be modulated by cholesterol and by bile acids that interact with the channel complex at distinct sites, including at the β1 subunits [37].
4. Conclusions and Perspectives
Cells of the immune system contain multiple GPCRs and ion channels that are modulated by oxysterols. However, in many cases, it is unclear if these signalling proteins are influenced by cholesterol oxidation products in vivo under physiological or pathological settings. Similarly, many of the consequences of oxysterol modulation of GPCRs and ion channels within the immune system have not been investigated. Furthermore, the molecular mechanisms by which oxysterols alter the function of many of these receptors awaits full characterization. Consequently, there is considerable scope for exploration of cell-surface receptors for oxysterols in the immune system, both for scientific discovery and for the development of new immunotherapeutic regimes.
References
- Kloudova, A.; Guengerich, F.P.; Soucek, P. The Role of Oxysterols in Human Cancer. Trends Endocrinol. Metab. 2017, 28, 485–496.
- Zmysłowski, A.; Szterk, A. Current knowledge on the mechanism of atherosclerosis and pro-atherosclerotic properties of oxysterols. Lipids Health Dis. 2017, 16, 188.
- Luu, W.; Sharpe, L.J.; Capell-Hattam, I.; Gelissen, I.C.; Brown, A.J. Oxysterols: Old Tale, New Twists. Annu. Rev. Pharm. Toxicol. 2016, 56, 447–467.
- Choi, C.; Finlay, D.K. Diverse Immunoregulatory Roles of Oxysterols—The Oxidized Cholesterol Metabolites. Metabolites 2020, 10, 384.
- Duc, D.; Vigne, S.; Pot, C. Oxysterols in Autoimmunity. Int. J. Mol. Sci. 2019, 20, 4522.
- Spann, N.J.; Glass, C.K. Sterols and oxysterols in immune cell function. Nat. Immunol. 2013, 14, 893–900.
- Ma, L.; Nelson, E.R. Oxysterols and nuclear receptors. Mol. Cell. Endocrinol. 2019, 484, 42–51.
- Korf, H.; Beken, S.V.; Romano, M.; Steffensen, K.; Stijlemans, B.; Gustafsson, J.; Grooten, J.; Huygen, K. Liver X receptors contribute to the protective immune response against Mycobacterium tuberculosis in mice. J. Clin. Investig. 2009, 119, 1626–1637.
- A DeBose-Boyd, R.; Brown, M.S.; Li, W.-P.; Nohturfft, A.; Goldstein, J.L.; Espenshade, P.J. Transport-Dependent Proteolysis of SREBP: Relocation of Site-1 Protease from Golgi to ER Obviates the Need for SREBP Transport to Golgi. Cell 1999, 99, 703–712.
- Gong, Y.; Lee, J.N.; Brown, M.S.; Goldstein, J.L.; Ye, J. Juxtamembranous aspartic acid in Insig-1 and Insig-2 is required for cholesterol homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 6154–6159.
- Repa, J.; Liang, G.; Ou, J.; Bashmakov, Y.; Lobaccaro, J.-M.A.; Shimomura, I.; Shan, B.; Brown, M.S.; Goldstein, J.L.; Mangelsdorf, D. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000, 14, 2819–2830.
- Liang, G.; Yang, J.; Horton, J.D.; Hammer, R.E.; Goldstein, J.L.; Brown, M.S. Diminished Hepatic Response to Fasting/Refeeding and Liver X Receptor Agonists in Mice with Selective Deficiency of Sterol Regulatory Element-binding Protein-1c. J. Biol. Chem. 2002, 277, 9520–9528.
- Shen, Z.-J.; Hu, J.; Kashi, V.P.; Kelly, E.; Denlinger, L.C.; Lutchman, K.; McDonald, J.G.; Jarjour, N.N.; Malter, J.S. Epstein-Barr Virus–induced Gene 2 Mediates Allergen-induced Leukocyte Migration into Airways. Am. J. Respir. Crit. Care Med. 2017, 195, 1576–1585.
- Emgård, J.; Kammoun, H.; García-Cassani, B.; Chesné, J.; Parigi, S.M.; Jacob, J.-M.; Cheng, H.-W.; Evren, E.; Das, S.; Czarnewski, P.; et al. Oxysterol Sensing through the Receptor GPR183 Promotes the Lymphoid-Tissue-Inducing Function of Innate Lymphoid Cells and Colonic Inflammation. Immunity 2018, 48, 120–132.
- Wyss, A.; Raselli, T.; Perkins, N.; Ruiz, F.; Schmelczer, G.; Klinke, G.; Moncsek, A.; Roth, R.; Spalinger, M.R.; Hering, L.; et al. The EBI2-oxysterol axis promotes the development of intestinal lymphoid structures and colitis. Mucosal Immunol. 2019, 12, 733–745.
- Yi, T.; Wang, X.; Kelly, L.M.; An, J.; Xu, Y.; Sailer, A.; Gustafsson, J.-A.; Russell, D.; Cyster, J.G. Oxysterol Gradient Generation by Lymphoid Stromal Cells Guides Activated B Cell Movement during Humoral Responses. Immunity 2012, 37, 535–548.
- Lu, E.; Dang, E.V.; McDonald, J.G.; Cyster, J.G. Distinct oxysterol requirements for positioning naïve and activated dendritic cells in the spleen. Sci. Immunol. 2017, 2, eaal5237.
- Raccosta, L.; Fontana, R.; Traversari, C.; Russo, V. Oxysterols recruit tumor-supporting neutrophils within the tumor microenvironment. OncoImmunology 2013, 2, e26469.
- Legler, D.F.; Matti, C.; Laufer, J.M.; Jakobs, B.D.; Purvanov, V.; Allmen, E.U.-V.; Thelen, M. Modulation of Chemokine Receptor Function by Cholesterol: New Prospects for Pharmacological Intervention. Mol. Pharm. 2017, 91, 331–338.
- Zhang, X.; Zhao, F.; Wu, Y.; Yang, J.; Han, G.W.; Zhao, S.; Ishchenko, A.; Ye, L.; Lin, X.; Ding, K.; et al. Crystal structure of a multi-domain human smoothened receptor in complex with a super stabilizing ligand. Nat. Commun. 2017, 8, 15383.
- Huang, P.; Zheng, S.; Wierbowski, B.; Kim, Y.; Nedelcu, D.; Aravena, L.; Liu, J.; Kruse, A.C.; Salic, A. Structural Basis of Smoothened Activation in Hedgehog Signaling. Cell 2018, 174, 312–324.
- Deshpande, I.; Liang, J.; Hedeen, D.; Roberts, K.J.; Zhang, Y.; Ha, B.; Latorraca, N.R.; Faust, B.; Dror, R.O.; Beachy, P.A.; et al. Smoothened stimulation by membrane sterols drives Hedgehog pathway activity. Nat. Cell Biol. 2019, 571, 284–288.
- Phelan, K.; Mahler, H. Acute Exposure to 25-Hydroxy-cholesterol Selectively Reduces GABAb and Not GABAa Receptor-Mediated Synaptic Inhibition. Biochem. Biophys. Res. Commun. 1997, 237, 68–73.
- A Kuhn, S.; van Landeghem, F.; Zacharias, R.; Färber, K.; Rappert, A.; Pavlovic, S.; Hoffmann, A.; Nolte, C.; Kettenmann, H. Microglia express GABAB receptors to modulate interleukin release. Mol. Cell. Neurosci. 2004, 25, 312–322.
- Huang, S.; Mao, J.; Wei, B.; Pei, G. The anti-spasticity drug baclofen alleviates collagen-induced arthritis and regulates dendritic cells. J. Cell. Physiol. 2015, 230, 1438–1447.
- Hammoud, Y.; Rice, T.; Mackrill, J.J. Oxysterols modulate calcium signalling in the A7r5 aortic smooth muscle cell-line. Biochimie 2013, 95, 568–577.
- Zhong, W.; Yi, Q.; Xu, B.; Li, S.; Wang, T.; Liu, F.; Zhu, B.; Hoffmann, P.R.; Ji, G.; Lei, P.; et al. ORP4L is essential for T-cell acute lymphoblastic leukemia cell survival. Nat. Commun. 2016, 7, 12702.
- Cao, X.; Chen, J.; Li, D.; Xie, P.; Xu, M.; Lin, W.; Li, S.; Pan, G.; Tang, Y.; Xu, J.; et al. ORP4L couples IP3to ITPR1 in control of endoplasmic reticulum calcium release. FASEB J. 2019, 33, 13852–13865.
- Boldyrev, A.A.; Carpenter, D.O.; Johnson, P. Emerging evidence for a similar role of glutamate receptors in the nervous and immune systems. J. Neurochem. 2005, 95, 913–918.
- Mashkina, A.P.; Tyulina, O.V.; Solovyova, T.I.; Kovalenko, E.; Kanevski, L.M.; Johnson, P.; Boldyrev, A.A. The excitotoxic effect of NMDA on human lymphocyte immune function. Neurochem. Int. 2007, 51, 356–360.
- Kahlfuß, S.; Simma, N.; Mankiewicz, J.; Bose, T.; Lowinus, T.; Klein-Hessling, S.; Sprengel, R.; Schraven, B.; Heine, M.; Bommhardt, U. Immunosuppression by N-Methyl-d-Aspartate Receptor Antagonists Is Mediated through Inhibition of Kv1.3 and KCa3.1 Channels in T Cells. Mol. Cell. Biol. 2013, 34, 820–831.
- Simma, N.; Bose, T.; Kahlfuß, S.; Mankiewicz, J.; Lowinus, T.; Lühder, F.; Schüler, T.; Schraven, B.; Heine, M.; Bommhardt, U. NMDA-receptor antagonists block B-cell function but foster IL-10 production in BCR/CD40-activated B cells. Cell Commun. Signal. 2014, 12, 75.
- Black, J.A.; Waxman, S.G. Noncanonical Roles of Voltage-Gated Sodium Channels. Neuron 2013, 80, 280–291.
- Roselli, F.; Livrea, P.; Jirillo, E. Voltage-Gated Sodium Channel Blockers as Immunomodulators. Recent Pat. CNS Drug Discov. 2006, 1, 83–91.
- Gonzalez-Perez, V.; Lingle, C.J. Regulation of BK Channels by Beta and Gamma Subunits. Annu. Rev. Physiol. 2019, 81, 113–137.
- Tian, Y.; Heinemann, S.H.; Hoshi, T. Large-conductance Ca2+- and voltage-gated K+channels form and break interactions with membrane lipids during each gating cycle. Proc. Natl. Acad. Sci. USA 2019, 116, 8591–8596.
- Dopico, A.M.; Bukiya, A.N. Regulation of Ca2+-Sensitive K+ Channels by Cholesterol and Bile Acids via Distinct Channel Subunits and Sites. Curr. Top. Membr. 2017, 80, 53–93.

