 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | HINDA NAJEM | + 2384 word(s) | 2384 | 2021-08-18 12:18:15 | | | |
| 2 | Beatrix Zheng | + 419 word(s) | 2803 | 2021-08-27 15:46:40 | | |
Video Upload Options
Gliomas are the most common primary brain tumors and are classified by the World Health Organization (WHO) as grade I–IV tumors based on molecular and genomic features, allowing a more accurate classification of patients. Given the transition from histological characterization, this newer molecular classification system enables precision medicine therapeutic selection and leads to a more accurate prediction of prognosis. Despite the evolution of such classifications and of immunotherapies, these tumors remain refractory to immune therapeutics. Understanding the microenvironment of gliomas and it's heterogeneity is key to discover potential future immunotherapeutic strategies. Recent findings show that immune activity against tumors may be dependent of the tumor microenvironment, especially in hot spots of immune reactivity like the invasive edge.
1. Introduction
Gliomas are the most common primary brain tumors and are classified by the World Health Organization (WHO) as grade I–IV tumors based on molecular and genomic features, allowing a more accurate classification of patients. Given the transition from histological characterization, this newer molecular classification system enables precision medicine therapeutic selection and leads to a more accurate prediction of prognosis [1][2]. One of the most central genetic characteristics is the isocitrate dehydrogenase mutation (IDH1) status that is commonly expressed in low-grade gliomas and reflects a favorable prognosis relative to IDH1 wild-type gliomas that are high-grade glioblastomas [3][4]. Glioblastoma is particularity challenging to treat despite multi-modal therapy, and median survival is 14.6 months [4][5].
Despite the beneficial effects of immunotherapies in multiple types of cancers including brain metastases from several solid tumors [6][7], the vast majority of glioma patients do not benefit. In fact, there is a lack in the understanding of glioma microenvironment that needs to be further elaborated in order to explain the hindering effects in the use of the available immunotherapies in the treatment of glioblastoma. Recent studies have shown that the immune composition and states are unique and specific to cancer lineage [8][9]. Additionally, there are differences in immune suppressive pathways and immune targets not only between lineages [10], but also between individual patients harboring the same types of cancers [11]. Further abrogating potential immune effector responses in gliomas, are a wide variety of immune suppressive secreted factors, exhaustion and sequestration of T cells in the bone marrow, administration of steroids, and the blood–brain barrier (BBB) that have been recently reviewed [12].
2. The Unique Immune Microenvironment of Gliomas
Gliomas, especially glioblastomas, are typically described as having a “cold” microenvironment given the paucity of TILs displaying an effector response. Molecularly distinct gliomas, depending on their IDH mutation status, have different immune compositions and landscape that defines its TME [13][14][15]. IDH-mutant gliomas are almost totally devoid of TILs in comparison with brain metastasis that are highly enriched with activated and exhausted T cells [8][16]. This status of low infiltration of T cells in gliomas, especially in the IDH-mutant subtype, creates an environment with low expression of immune checkpoint targets, which provides one possible reason for the resistance to immune checkpoint inhibitors. On the other hand, IDH-mutant tumors are enriched with tumor-resident microglia, which is in contrast to IDH wild-type and brain metastasis that are infiltrated with monocyte-derived macrophages originating from the periphery [8][15][16]. In contrast to brain metastasis, 40% to 70% of the total immune cell populations in glioblastoma are myeloid derived cells, representing the most abundant immune cell type infiltrating these tumors [17] that typically have an immune suppressive phenotype.
In addition to the different immune populations that populate different cancer lineages within the CNS, unbiased transcriptional profiling is beginning to reveal distinct subpopulations and states that further increase immunological complexity and heterogeneity even within the same immune cell lineage in the TME. Based on transcriptomics, there may be many different microglia subtypes that demonstrate different functional signatures such as phagocytosis, antigen presentation and lytic functions [18]. Although conventional described in the context of M1 (tumor suppressive/immune supportive) or M2 (tumor supportive/immune suppressive) phenotypes, ex vivo data of gliomas have revealed that neither of these were adequate descriptions for tumor associated macrophages (TAMs) [19]. With emerging single cell sequencing data, distinct subtypes/subgroups will be emerging that were not evident with bulk profiling initiatives. The innate immune cells in gliomas likely express vastly different molecular phenotypes, transcriptional states and functionalities that are influenced by cancer lineage (gliomas or metastasis), genetic status (IDH-mutation status), and within the TME location (hypoxia) further confounding the heterogeneity of the TME [19][20] likely even within the same tumor. This area needs further investigation, including functional immune characterization, in order to comprehend the different subtypes of the various tumor associated immune cells and to be fully informed regarding therapeutic selection and optimization.
Additional evidence that the glioma TME is distinct from other cancer lineages is the lack of tumor mutational burden (TMB) being a predictive response biomarker to immune checkpoint inhibitors. McGrail et al. recently showed in an analysis of over 10,000 patients that not all tumors with high-TMB developed effector immune responses to immune checkpoint inhibitors therapy [21]. Specifically, in breast, prostate, and glioma cancer patients there appears to be no correlation between CD8 T cell levels and neoantigen load, with overall survival [22]. In another analysis of recurrent glioblastoma patients, a very low-TMB state was associated with better response to immune checkpoint inhibition or polio virotherapy [23]. The distinction may reside in the mutational composition found in glioma not being particularly immunogenic or alternatively that the mutagenic antigens are eliminated potentially due to better immune surveillance in the patients with lower TMB, resulting in better immune recognition and elimination of mutated sub-clones before the initiation of immunotherapy. Such immune environment would keep the overall TMB low and would explain the otherwise apparent discordance between studies of patients with constitutional DNA mismatch repair syndromes that show responses to immunotherapy. Other investigators have shown that specific mutations like PTEN, are associated with worse outcomes to immune checkpoint inhibitors and one could consider removing these subjects from use of immune checkpoint inhibitors [24], rather than enrichment selection based on TMB. In contrast, in recurrent glioblastoma patients that respond to PD-1 blockade, there is enriched expression of BRAF and PTPN11 activation mutations [24][25]. Arietta et al. showed that phosphorylated ERK1/2 (p-ERK1/2) proteins are predictors of response in recurrent glioblastoma patients to PD-1 inhibition even in absence of BRAF and PTPN11 expression [26]. Given the complexity of the steps needed to mount an effective anti-tumor immune response, it is likely that prediction of responsiveness to immunotherapies will not be predicated on a single marker. A systems biology approach possibly using different analysis platforms, through genome sequencing, emerging current single cell analysis, immune landscape observation and functional analysis may need to be consolidated to ultimately predict responses and ultimately understand this unique microenvironment.
3. Differential T Cell Deactivation and Suppression in CNS Cancer Lineages
Multiple immune suppressive mechanisms and pathways are utilized by the tumor cells and their environment to inhibit anti-tumor eradication. T cells are usually the effectors of the cytotoxic anti-tumor immune response in many cancers and if found in the glioblastoma TME, they are under a dysfunctional state and exhausted. Exhaustion occurs after repeated antigen exposure that leads to the expression of multiple immune checkpoints on the T cell surface [27]. Brain metastases are more enriched with TILs relative to glioblastoma that have abundant tumor-associated myeloid cells (TAMs) [8][16]. As such, engagement of immune checkpoints and modulation is more available for targeting in brain metastasis relative to glioblastoma. Notably, the expression of TIM-3 and LAG-3 immune checkpoints is relatively low in gliomas [11], and as such these therapeutic targets are unlikely to be beneficial for the treatment of patients with glioma for most patients and especially as a monotherapy. Both cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed death 1 (PD-1), that are induced in activated T cells providing inhibitory signals by binding to their ligands expressed on the surface of antigen presenting cells or tumor cells ( Figure 1 ), are more frequently expressed. Multiple clinical trials have shown that brain metastases are more responsive to immune checkpoint inhibitors therapies such anti-PD-1 and anti-CTLA-4 [6][28][29] relative to glioblastoma [30][31]. Other immune checkpoints, like TIGIT, were identified to be highly expressed on T cells, especially cytotoxic CD8 + tumor infiltrating T cells in murine models of glioma [32]. More recently, through an analysis of glioma infiltrating T cells, Mathewson et al. found that these cells express natural killer (NK) genes. They demonstrated an enhanced anti-tumor effect of these T cells through the direct blockade of CD161, a NK cell receptor expressed on the T cells surface, or by the inactivation of its respective NK gene KLRB1, highlighting the role of CD161 and potential therapeutic intervention [33]. The association of all these immunosuppressive markers on T cells surface induce a state of reduced responsiveness to immune stimulations and thus inhibited effector immune response.
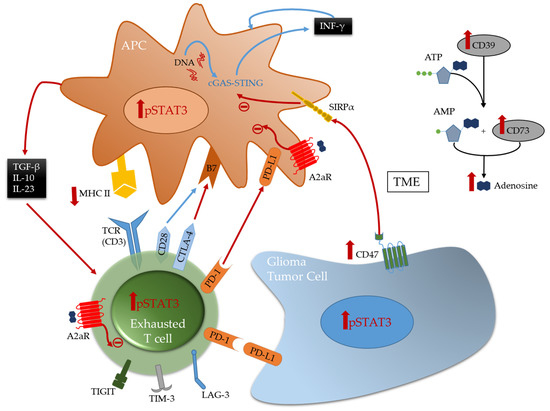
A frequently expressed immune suppressive pathway in glioblastoma is the A2aR-adenosine pathway [34][35]. Related to this pathway is CD73, an enzyme shown to be preferentially expressed by tumor cells in glioma patients [10] and upregulated in immune cells [11], especially in IDH1-mutant gliomas. This ectonucleotidase is responsible for the activation of adenosine that binds to the A2a receptor on both T cells and myeloid cells thereby triggering immunosuppression [34] ( Figure 1 ). Therefore, this pathway is a potential therapeutic target. However, only limited therapeutic benefits were shown by administration of adenosine receptor inhibitors in multiple murine models of gliomas, including those expressing CD73 even in combination with other inhibitors [11]. This highlights the challenges in approaches aimed to reverse T cell exhaustion induced in glioma TME [32][36].
4. Emerging Strategies for Modulation of Immunosuppressive Tumor Associated Macrophages
TAMs, the most abundant immune cells present in the TME, express a wide variety of immunosuppressive phenotypes [37]. Gliomas express CD47 which blocks phagocytosis thereby evading immune recognition and eradication [38][39]. CD47 blockade in several murine models [38][40] has shown therapeutic benefit by freeing the innate immune system to activate APCs thereby leading to effector CD8 + effector T cells mediated immune responses [41][42]. However, monotherapy leads to treatment resistance [42][43][44]. Therefore, combination therapies can represent a solution for better induced anti-tumor responses and outcomes. A recent study demonstrated that combining anti-CD47 targeted therapy with temozolomide creates a pro-phagocytosis effect and induces antigen presentation by activating and upregulating the cGAS-STING pathway in APCs, thus leading to an effective adaptive immune response [44].
Stimulator of interferon genes (STING) is an important component of the innate immune response to pathogenic DNA. It is a widely expressed sensor of cellular stress, activated by cytosolic cyclic dinucleotides, which may be released by bacteria or created through cytosolic self- or viral-DNA interaction with cyclic GMP-AMP synthase (cGAS) [45][46]. STING agonists can induce T cell infiltration into tumors known to be devoid of such immune composition and in tumors enriched with myeloid immune cells through pro-inflammatory activation resulting in marked in vivo therapeutic activity [47][48]. This pathway bridges the innate and adaptive immune systems both by triggering interferon (IFN) release and through activation of myeloid cells ( Figure 1 ). Distinct from most other innate immune agonists, STING activation can re-educate tumor-supportive immunosuppressive macrophages toward a pro-inflammatory phenotype and can reverse the suppressive phenotype of myeloid-derived suppressor cells (MDSCs) [49][50]. STING agonists have demonstrated radiographic responses in canines with high-grade gliomas [51]. In summary, STING agonists may be a compelling therapeutic strategy for gliomas because they: (1) can simulate a foreign body reaction, thus providing a “target”; (2) induce IFN, thereby providing potent T cell effector action; (3) induce chemokine production and thus T cell trafficking to the tumor; and they are (4) easy to synthesize.
The reciprocal immune modulatory strategy of TAMs is to block a key deactivating pathway the signal transducer and activator of transcription 3 (STAT3). STAT3 is an important mechanism of suppression of both innate and adaptive components of the immune system ( Figure 2 ). STAT3 expression shifts the TAMs to an immunosuppressive phenotype secreting immunomodulatory suppressive factors like IL-10 and TGF-β, and impairing phagocytosis and antigen presentation [52][53][54]. Similarly, STAT3 impairs maturation and antigen presentation by dendritic cells (DCs) preventing T cell activation and proliferation [55][56]. The inhibition of STAT3 can reactivate the immune system in the TME by promoting infiltrating DCs maturation, increasing expression of the co-stimulatory molecules (CD80/CD86) necessary for T cell activation, and decreasing the number of myeloid derived suppressor cells (MDSCs) in the immune microenvironment [57][58][59]. Additionally, STAT3 is a key inducer of immune suppressive cytokines ( IL-10, IL-4, IL-6 and TGF-β), maintains immunosuppressive cell cross-talk [60][61][62][63][64], increases tumor infiltration by MDSCs, and induces T cell arrest and apoptosis [65][66][67][68]. Through MDSCs secretion of INF-α and other mechanisms, STAT3 upregulates the expression of inhibitory immune checkpoints like PD-L1 on the surface of TAMs and tumor-infiltrating DCs [62][66]. Furthermore, STAT3 signaling is correlated with a decrease in effector T cells infiltration of the tumor and prevents CD8 + T cell activation by increasing the secretion of INF-γ [69]. STAT3 activation correlates with the activation and upregulation of FOXP3 in T cells and is a key inducer of immunosuppressive Tregs infiltration of the TME [70][71][72][73]. Several studies, in mice models of glioma and on patient-extracted glioma tumor cells, have demonstrated the benefits of inhibiting STAT3 that enhances the anti-tumor immune response by improving T cell, DCs and NK activation in the TME [55][74].
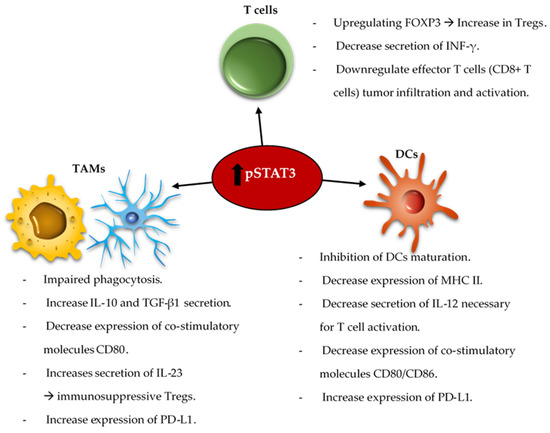
The combination of the standard of care radiation therapy with a STAT3 inhibitor, showed improvement in the overall survival of mice harboring intracranial gliomas [75]. This was shown to be immunologically mediated with reprogramming of the DCs in the TME. STAT3 signaling is involved in a variety of intrinsic and acquired resistance mechanisms to administered anti-tumor therapies such as temozolomide [76][77][78], radiation [79][80][81][82][83][84][85], and targeted therapies [86][87][88]. As such, some studies demonstrated that the combination of anti-VEGF (Cediranib) and STAT3 inhibitors (AZD1480) for the treatment of glioblastoma in a mice model led to a decrease in the tumor volume and angiogenesis [89][90]. The co-inhibition of STAT3 and MET induce glioma tumor cells destruction by reactivating apoptosis mechanisms [91]. As such, an area of future investigation is combinatorial immune modulatory or chemotherapeutic strategies with STAT3 inhibition.
5. Conclusions
References
- Komori, T. The 2016 WHO Classification of Tumours of the Central Nervous System: The Major Points of Revision. Neurol. Med. Chir. 2017, 57, 301–311.
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820.
- Brat, D.J.; Aldape, K.; Colman, H.; Figrarella-Branger, D.; Fuller, G.N.; Giannini, C.; Holland, E.C.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.; Komori, T. cIMPACT-NOW update 5: Recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol. 2020, 139, 603–608.
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100.
- Stupp, R.; Mason, W.P.; Bent, M.V.D.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. New Engl. J. Med. 2005, 352, 987–996.
- Tawbi, H.A.; Forsyth, P.A.; Algazi, A.; Hamid, O.; Hodi, F.S.; Moschos, S.J.; Khushalani, N.I.; Lewis, K.; Lao, C.D.; Postow, M.A.; et al. Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. New Engl. J. Med. 2018, 379, 722–730.
- Hendriks, L.E.; Henon, C.; Auclin, E.; Mezquita, L.; Ferrara, R.; Audigier-Valette, C.; Mazieres, J.; Lefebvre, C.; Rabeau, A.; Le Moulec, S.; et al. Outcome of Patients with Non–Small Cell Lung Cancer and Brain Metastases Treated with Checkpoint Inhibitors. J. Thorac. Oncol. 2019, 14, 1244–1254.
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20.
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341.
- Goswami, S.; Walle, T.; Cornish, A.E.; Basu, S.; Anandhan, S.; Fernandez, I.; Vence, L.; Blando, J.; Zhao, H.; Yadav, S.S.; et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med. 2019, 26, 39–46.
- Ott, M.; Tomaszowski, K.-H.; Marisetty, A.; Kong, L.-Y.; Wei, J.; Duna, M.; Blumberg, K.; Ji, X.; Jacobs, C.; Fuller, G.N.; et al. Profiling of patients with glioma reveals the dominant immunosuppressive axis is refractory to immune function restoration. JCI Insight 2020, 5e134386.
- Chuntova, P.; Chow, F.; Watchmaker, P.B.; Galvez, M.; Heimberger, A.B.; Newell, E.W.; Diaz, A.; DePinho, R.A.; Li, M.O.; Wherry, E.J. Unique challenges for glioblastoma immunotherapy—discussions across neuro-oncology and non-neuro-oncology experts in cancer immunology. Meeting Report from the 2019 SNO Immuno-Oncology Think Tank. Neuro Oncol. 2021, 23, 356–375.
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Investig. 2017, 127, 1425–1437.
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203.
- Friedrich, M.; Sankowski, R.; Bunse, L.; Kilian, M.; Green, E.; Guevara, C.R.; Pusch, S.; Poschet, G.; Sanghvi, K.; Hahn, M.; et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat. Rev. Cancer 2021, 2, 723–740.
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17.
- Hussain, S.F.; Yang, D.; Suki, D.; Grimm, E.; Heimberger, A.B. Innate immune functions of microglia isolated from human glioma patients. J. Transl. Med. 2006, 4, 15.
- Gupta, P.; Dang, M.; Bojja, K.; Shehwana, H.; Tran, T.; Wang, L.; Bhat, K. 540 Transcriptionally defined immune landscape in human gliomas. J. Immunother. Cancer 2020, 8, A576.
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1e85841.
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macro-phage activation in the tumor microenvironment. Genome. Biol. 2017, 18, 234.
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nat. Cell Biol. 2020, 580, 517–523.
- McGrail, D.; Pilié, P.; Rashid, N.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.; Lim, B.; et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 2021, 32, 661–672.
- Gromeier, M.; Brown, M.C.; Zhang, G.; Lin, X.; Chen, Y.; Wei, Z.; Beaubier, N.; Yan, H.; He, Y.; Desjardins, A.; et al. Very low mutation burden is a feature of inflamed recurrent glioblastomas responsive to cancer immunotherapy. Nat. Commun. 2021, 12, 1–7.
- Zhao, J.; Chen, A.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469.
- Lukas, R.V.; Rodon, J.; Becker, K.; Wong, E.T.; Shih, K.; Touat, M.; Fassò, M.; Osborne, S.; Molinero, L.; O’Hear, C.; et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J. Neuro Oncol. 2018, 140, 317–328.
- Arrieta, V.A.; Chen, A.X.; Kane, J.R.; Kang, S.J.; Kassab, C.; Dmello, C.; Zhao, J.; Burdet, K.B.; Upadhyayula, P.; Lee-Chang, C.; et al. ERK1/2 phosphorylation predicts survival following anti-PD-1 immunotherapy in recurrent glioblastoma. Nature Cancer. 2021; In Press.
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221.
- Tawbi, H.A.-H.; Forsyth, P.A.; Hodi, F.S.; Lao, C.D.; Moschos, S.J.; Hamid, O.; Atkins, M.B.; Lewis, K.D.; Thomas, R.P.; Glaspy, J.A. Efficacy and safety of the combination of nivolumab (NIVO) plus ipilimumab (IPI) in patients with symptomatic melanoma brain metastases (CheckMate 204). Am. Soc. Clin. Oncol. 2019, 37, 9501.
- Goldberg, S.B.; Schalper, K.A.; Gettinger, S.N.; Mahajan, A.; Herbst, R.S.; Chiang, A.C.; Lilenbaum, R.; Wilson, F.H.; Omay, S.B.; James, B.Y. Pembrolizumab for management of patients with NSCLC and brain metastases: Long-term results and biomarker analysis from a non-randomised, open-label, phase 2 trial. The Lancet Oncology 2020, 21, 655–663.
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: The CheckMate 143 phase 3 randomized clinical trial. JAMA oncology 2020, 6, 1003–1010.
- Reardon, D.A.; Omuro, A.; Brandes, A.A.; Rieger, J.; Wick, A.; Sepulveda, J.; Phuphanich, S.; De Souza, P.; Ahluwalia, M.S.; Lim, M.; et al. OS10.3 Randomized Phase 3 Study Evaluating the Efficacy and Safety of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: CheckMate 143. Neuro-Oncology 2017, 19, iii21.
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; DeChant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186.
- Mathewson, N.D.; Ashenberg, O.; Tirosh, I.; Gritsch, S.; Perez, E.M.; Marx, S.; Jerby-Arnon, L.; Chanoch-Myers, R.; Hara, T.; Richman, A.R.; et al. Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell 2021, 184, 1281–1298.e26.
- Leone, R.D.; Lo, Y.-C.; Powell, J.D. A2aR antagonists: Next generation checkpoint blockade for cancer immunotherapy. Comput. Struct. Biotechnol. J. 2015, 13, 265–272.
- Mohan, A.A.; Tomaszewski, W.H.; Haskell-Mendoza, A.P.; Hotchkiss, K.M.; Singh, K.; Reedy, J.L.; Fecci, P.E.; Sampson, J.H.; Khasraw, M. Targeting Immunometabolism in Glioblastoma. Front. Oncol. 2021, 11.
- Ott, M.; Prins, R.M.; Heimberger, A.B. The immune landscape of common CNS malignancies: Implications for immunotherapy. Nat. Rev. Clin. Oncol. 2021, 1–16.
- De Groot, J.; Penas-Prado, M.; Alfaro-Munoz, K.; Hunter, K.; Pei, B.L.; O’Brien, B.; Weathers, S.-P.; Loghin, M.; Kamiya Matsouka, C.; Yung, W.A. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro Oncol. 2020, 22, 539–549.
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.; Van Rooijen, N.; Weissman, I.L. CD47 Is an Adverse Prognostic Factor and Therapeutic Antibody Target on Human Acute Myeloid Leukemia Stem Cells. Cell 2009, 138, 286–299.
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 Antibody Synergizes with Rituximab to Promote Phagocytosis and Eradicate Non-Hodgkin Lymphoma. Cell 2010, 142, 699–713.
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Jan, M.; Weissman-Tsukamoto, R.; Zhao, F.; Park, C.Y.; Weissman, I.L.; Majeti, R. Therapeutic Antibody Targeting of CD47 Eliminates Human Acute Lymphoblastic Leukemia. Cancer Res. 2010, 71, 1374–1384.
- Tseng, D.; Volkmer, J.-P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M.; et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103–11108.
- Liu, X.; Pu, Y.; Cron, K.R.; Deng, L.; Kline, J.; Frazier, W.A.; Xu, H.; Peng, H.; Fu, Y.-X.; Xu, M.M. CD47 blockade triggers T cell–mediated destruction of immunogenic tumors. Nat. Med. 2015, 21, 1209–1215.
- Horrigan, S.K.; Iorns, E. Replication Study: The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. eLife 2017, 6, e18173.
- von Roemeling, C.A.; Wang, Y.; Qie, Y.; Yuan, H.; Zhao, H.; Liu, X.; Yang, Z.; Yang, M.; Deng, W.; Bruno, K.A. Therapeutic modulation of phagocytosis in glioblastoma can activate both innate and adaptive antitumour immunity. Nat. Commun. 2020, 11, 1–12.
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Sci. 2012, 339, 786–791.
- Burdette, D.L.; Monroe, K.M.; Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING is a direct innate immune sensor of cyclic di-GMP. Nat. Cell Biol. 2011, 478, 515–518.
- Ohkuri, T.; Ghosh, A.; Kosaka, A.; Zhu, J.; Ikeura, M.; David, M.; Watkins, S.C.; Sarkar, S.N.; Okada, H. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol. Res. 2014, 2, 1199–1208.
- Ohkuri, T.; Ghosh, A.; Kosaka, A.; Sarkar, S.; Okada, H. Protective role of STING against gliomagenesis: Rational use of STING agonist in anti-glioma immunotherapy. OncoImmunology 2015, 4, e999523.
- Da Hoong, B.Y.; Gan, Y.H.; Liu, H.; Chen, E.S. cGAS-STING pathway in oncogenesis and cancer therapeutics. Oncotarget 2020, 11, 2930.
- Wan, D.; Jiang, W.; Hao, J. Research advances in how the cGAS-STING pathway controls the cellular inflammatory response. Front. Immunol. 2020, 11, 615.
- Boudreau, C.E.; Najem, H.; Ott, M.; Horbinski, C.; Fang, D.; DeRay, C.M.; Levine, J.; Curran, M.; Heimberger, A.B. Intratumoral delivery of STING agonist results in radiographic response in canine glioblastoma. In Proceedings of the AACR Annual Meeting 2021, Philadelphia, PA, USA, 10–15 April 2021. Submitted.
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage M1–M2 polarization balance. Front. Immunol. 2014, 5, 614.
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440.
- Wu, A.; Wei, J.; Kong, L.-Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro-Oncology 2010, 12, 1113–1125.
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mulé, J.; Kerr, W.; et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321.
- Molavi, O.; Ma, Z.; Hamdy, S.; Lavasanifar, A.; Samuel, J. Immunomodulatory and anticancer effects of intra-tumoral co-delivery of synthetic lipid A adjuvant and STAT3 inhibitor, JSI-124. Immunopharmacol. Immunotoxicol. 2008, 31, 214–221.
- Fujita, M.; Zhu, X.; Sasaki, K.; Ueda, R.; Low, K.L.; Pollack, I.F.; Okada, H. Inhibition of STAT3 Promotes the Efficacy of Adoptive Transfer Therapy Using Type-1 CTLs by Modulation of the Immunological Microenvironment in a Murine Intracranial Glioma. J. Immunol. 2008, 180, 2089–2098.
- Farren, M.R.; Carlson, L.M.; Netherby, C.S.; Lindner, I.; Li, P.-K.; Gabrilovich, D.I.; Abrams, S.I.; Lee, K.P. Tumor-induced STAT3 signaling in myeloid cells impairs dendritic cell generation by decreasing PKCβII abundance. Sci. Signal. 2014, 7, ra16.
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 Feed-Forward Loop Drives Tumorigenesis and Metastasis. Neoplasia 2013, 15, 848–862.
- Heiland, D.H.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.; Strähle, J.; Heynckes, S.; Grauvogel, J. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 2019, 10, 1–12.
- Chuang, H.-Y.; Su, Y.-K.; Liu, H.-W.; Chen, C.-H.; Chiu, S.-C.; Cho, D.-Y.; Lin, S.-Z.; Chen, Y.-S.; Lin, C.-M. Preclinical Evidence of STAT3 Inhibitor Pacritinib Overcoming Temozolomide Resistance via Downregulating miR-21-Enriched Exosomes from M2 Glioblastoma-Associated Macrophages. J. Clin. Med. 2019, 8, 959.
- Yao, Y.; Ye, H.; Qi, Z.; Mo, L.; Yue, Q.; Baral, A.; Hoon, D.S.; Vera, J.C.; Heiss, J.D.; Chen, C.C.; et al. B7-H4(B7x)–Mediated Cross-talk between Glioma-Initiating Cells and Macrophages via the IL6/JAK/STAT3 Pathway Lead to Poor Prognosis in Glioma Patients. Clin. Cancer Res. 2016, 22, 2778–2790.
- Li, J.; Shen, J.; Wang, Z.; Xu, H.; Wang, Q.; Chai, S.; Fu, P.; Huang, T.; Anas, O.; Zhao, H. ELTD1 facilitates glioma proliferation, migration and invasion by activating JAK/STAT3/HIF-1α signaling axis. Sci. Rep. 2019, 9, 1–12.
- Ko, H.-J.; Kim, Y.-J. Signal transducer and activator of transcription proteins: Regulators of myeloid-derived suppressor cell-mediated immunosuppression in cancer. Arch. Pharmacal Res. 2016, 39, 1597–1608.
- Poholek, C.H.; Raphael, I.; Wu, D.; Revu, S.; Rittenhouse, N.; Uche, U.U.; Majumder, S.; Kane, L.P.; Poholek, A.C.; McGeachy, M.J. Noncanonical STAT3 activity sustains pathogenic Th17 proliferation and cytokine response to antigen. J. Exp. Med. 2020, 217, e20191761.
- Xiao, W.; Klement, J.D.; Lu, C.; Ibrahim, M.L.; Liu, K. IFNAR1 Controls Autocrine Type I IFN Regulation of PD-L1 Expression in Myeloid-Derived Suppressor Cells. J. Immunol. 2018, 201, 264–277.
- Rajappa, P.; Cobb, W.S.; Vartanian, E.; Huang, Y.; Daly, L.; Hoffman, C.; Zhang, J.; Shen, B.; Yanowitch, R.; Garg, K.; et al. Malignant Astrocytic Tumor Progression Potentiated by JAK-mediated Recruitment of Myeloid Cells. Clin. Cancer Res. 2016, 23, 3109–3119.
- Vasquez-Dunddel, D.; Pan, F.; Zeng, Q.; Gorbounov, M.; Albesiano, E.; Fu, J.; Blosser, R.L.; Tam, A.J.; Bruno, T.; Zhang, H.; et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J. Clin. Investig. 2013, 123, 1580–1589.
- Yue, C.; Shen, S.; Deng, J.; Priceman, S.J.; Li, W.; Huang, A.; Yu, H. STAT3 in CD8+ T Cells Inhibits Their Tumor Accumulation by Downregulating CXCR3/CXCL10 Axis. Cancer Immunol. Res. 2015, 3, 864–870.
- El Andaloussi, A.; Lesniak, M.S. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme1. Neuro Oncol. 2006, 8, 234–243.
- Zorn, E.; Nelson, E.A.; Mohseni, M.; Porcheray, F.; Kim, H.; Litsa, D.; Bellucci, R.; Raderschall, E.; Canning, C.; Soiffer, R.J.; et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 2006, 108, 1571–1579.
- Pallandre, J.-R.; Brillard, E.; Créhange, G.; Radlovic, A.; Remy-Martin, J.-P.; Saas, P.; Rohrlich, P.S.; Pivot, X.; Ling, X.; Tiberghien, P.; et al. Role of STAT3 in CD4+CD25+FOXP3+Regulatory Lymphocyte Generation: Implications in Graft-versus-Host Disease and Antitumor Immunity. J. Immunol. 2007, 179, 7593–7604.
- Wei, J.; Wu, A.; Kong, L.-Y.; Wang, Y.; Fuller, G.; Fokt, I.; Melillo, G.; Priebe, W.; Heimberger, A.B. Hypoxia Potentiates Glioma-Mediated Immunosuppression. PLoS ONE 2011, 6, e16195.
- Hussain, S.F.; Kong, L.-Y.; Jordan, J.; Conrad, C.; Madden, T.; Fokt, I.; Priebe, W.; Heimberger, A.B. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007, 67, 9630–9636.
- Rath, B.H.; Wahba, A.; Camphausen, K.; Tofilon, P.J. Coculture with astrocytes reduces the radiosensitivity of glioblastoma stem-like cells and identifies additional targets for radiosensitization. Cancer Med. 2015, 4, 1705–1716.
- Kohsaka, S.; Wang, L.; Yachi, K.; Mahabir, R.; Narita, T.; Itoh, T.; Tanino, M.; Kimura, T.; Nishihara, H.; Tanaka, S. STAT3 Inhibition Overcomes Temozolomide Resistance in Glioblastoma by Downregulating MGMT Expression. Mol. Cancer Ther. 2012, 11, 1289–1299.
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Grogan, P.T.; Lamont, J.D.; Decker, P.A.; Wu, W.; James, C.D.; Sarkaria, J.N. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro-Oncology 2009, 11, 281–291.
- Li, H.; Chen, L.; Li, J.-J.; Zhou, Q.; Huang, A.; Liu, W.-W.; Wang, K.; Gao, L.; Qi, S.-T.; Lu, Y.-T. miR-519a enhances chemosensitivity and promotes autophagy in glioblastoma by targeting STAT3/Bcl2 signaling pathway. J. Hematol. Oncol. 2018, 11, 1–16.
- Yu, H.; Zhang, S.; Ibrahim, A.N.; Wang, J.; Deng, Z.; Wang, M. RCC2 promotes proliferation and radio-resistance in glioblastoma via activating transcription of DNMT1. Biochem. Biophys. Res. Commun. 2019, 516, 999–1006.
- Mrowczynski, O.D.; Madhankumar, A.B.; Sundstrom, J.; Zhao, Y.; Kawasawa, Y.I.; Slagle-Webb, B.; Mau, C.; Payne, R.A.; Rizk, E.B.; Zacharia, B.; et al. Exosomes impact survival to radiation exposure in cell line models of nervous system cancer. Oncotarget 2018, 9, 36083–36101.
- Li, C.; Ran, H.; Song, S.; Liu, W.; Zou, W.; Jiang, B.; Zhao, H.; Shao, B. Overexpression of RPN2 suppresses radiosensitivity of glioma cells by activating STAT3 signal transduction. Mol. Med. 2020, 26, 1–9.
- Ventero, M.P. Radiotherapy resistance acquisition in Glioblastoma. Role of SOCS1 and SOCS3. PLoS ONE 2019, 14, e0212581.
- Maachani, U.B.; Shankavaram, U.; Kramp, T.; Tofilon, P.J.; Camphausen, K.; Tandle, A.T. FOXM1 and STAT3 interaction confers radioresistance in glioblastoma cells. Oncotarget 2016, 7, 77365–77377.
- Zhong, C.; Tao, B.; Chen, Y.; Guo, Z.; Yang, X.; Peng, L.; Xia, X.; Chen, L. B7-H3 regulates glioma growth and cell invasion through a JAK2/STAT3/slug-dependent signaling pathway. OncoTargets Ther. 2020, 13, 2215.
- Lin, J.-C.; Tsai, J.-T.; Chao, T.-Y.; Ma, H.-I.; Liu, W.-H. The STAT3/Slug axis enhances radiation-induced tumor invasion and cancer stem-like properties in radioresistant glioblastoma. Cancers 2018, 10, 512.
- Network, C.G.A.R. Correction: Corrigendum: Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2013, 494, 506.
- Robe, P.A.; Martin, D.H.; Nguyen-Khac, M.T.; Artesi, M.; Deprez, M.; Albert, A.; Vanbelle, S.; Califice, S.; Bredel, M.; Bours, V. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of Sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 2009, 9, 372.
- Atkinson, G.P.; Nozell, S.E.; Benveniste, E.N. NF-κB and STAT3 signaling in glioma: Targets for future therapies. Expert Rev. Neurother. 2010, 10, 575–586.
- de Groot, J.; Liang, J.; Kong, L.-Y.; Wei, J.; Piao, Y.; Fuller, G.; Qiao, W.; Heimberger, A.B. Modulating antiangiogenic resistance by inhibiting the signal transducer and activator of transcription 3 pathway in glioblastoma. Oncotarget 2012, 3, 1036.
- Batchelor, T.T.; Reardon, D.A.; De Groot, J.F.; Wick, W.; Weller, M. Antiangiogenic therapy for glioblastoma: Current status and future prospects. Clin. Cancer Res. 2014, 20, 5612–5619.
- Cruickshanks, N.; Zhang, Y.; Hine, S.; Gibert, M.; Yuan, F.; Oxford, M.; Grello, C.M.; Pahuski, M.; Dube, C.; Guessous, F.; et al. Discovery and Therapeutic Exploitation of Mechanisms of Resistance to MET Inhibitors in Glioblastoma. Clin. Cancer Res. 2018, 25, 663–673.
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486.
- Jackson, C.M.; Kochel, C.M.; Nirschl, C.J.; Durham, N.M.; Ruzevick, J.; Alme, A.; Francica, B.J.; Elias, J.; Daniels, A.; Dubensky, T.W.; et al. Systemic Tolerance Mediated by Melanoma Brain Tumors Is Reversible by Radiotherapy and Vaccination. Clin. Cancer Res. 2015, 22, 1161–1172.
- Vonderheide, R.H. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell 2018, 33, 563–569.
- Gnjatic, S.; Sawhney, N.B.; Bhardwaj, N. Toll-like receptor agonists: Are they good adjuvants? Cancer J. 2010, 16, 382–391.
- Weichselbaum, R.R. Radiotherapy and immunotherapy: A beneficial liaison? Nat. Rev. Clin. Oncol. 2017, 14, 365–379.

