Gliomas are the most common primary brain tumors and are classified by the World Health Organization (WHO) as grade I–IV tumors based on molecular and genomic features, allowing a more accurate classification of patients. Given the transition from histological characterization, this newer molecular classification system enables precision medicine therapeutic selection and leads to a more accurate prediction of prognosis. Despite the evolution of such classifications and of immunotherapies, these tumors remain refractory to immune therapeutics. Understanding the microenvironment of gliomas and it's heterogeneity is key to discover potential future immunotherapeutic strategies. Recent findings show that immune activity against tumors may be dependent of the tumor microenvironment, especially in hot spots of immune reactivity like the invasive edge.
- glioma
- CNS metastasis
- immune composition
- tumor microenvironment
- immune therapy
- immune checkpoints
- T cells
- tumor associated macrophages/microglia
1. Introduction
Gliomas are the most common primary brain tumors and are classified by the World Health Organization (WHO) as grade I–IV tumors based on molecular and genomic features, allowing a more accurate classification of patients. Given the transition from histological characterization, this newer molecular classification system enables precision medicine therapeutic selection and leads to a more accurate prediction of prognosis [1][2][1,2]. One of the most central genetic characteristics is the isocitrate dehydrogenase mutation (IDH1) status that is commonly expressed in low-grade gliomas and reflects a favorable prognosis relative to IDH1 wild-type gliomas that are high-grade glioblastomas [3][4][3,4]. Glioblastoma is particularity challenging to treat despite multi-modal therapy, and median survival is 14.6 months [4][5][4,5].
Despite the beneficial effects of immunotherapies in multiple types of cancers including brain metastases from several solid tumors [6][7][6,7], the vast majority of glioma patients do not benefit. In fact, there is a lack in the understanding of glioma microenvironment that needs to be further elaborated in order to explain the hindering effects in the use of the available immunotherapies in the treatment of glioblastoma. Recent studies have shown that the immune composition and states are unique and specific to cancer lineage [8][9][8,9]. Additionally, there are differences in immune suppressive pathways and immune targets not only between lineages [10], but also between individual patients harboring the same types of cancers [11]. Further abrogating potential immune effector responses in gliomas, are a wide variety of immune suppressive secreted factors, exhaustion and sequestration of T cells in the bone marrow, administration of steroids, and the blood–brain barrier (BBB) that have been recently reviewed [12].
2. The Unique Immune Microenvironment of Gliomas
Gliomas, especially glioblastomas, are typically described as having a “cold” microenvironment given the paucity of TILs displaying an effector response. Molecularly distinct gliomas, depending on their IDH mutation status, have different immune compositions and landscape that defines its TME [13][14][15][24,25,26]. IDH-mutant gliomas are almost totally devoid of TILs in comparison with brain metastasis that are highly enriched with activated and exhausted T cells [8][16][8,27]. This status of low infiltration of T cells in gliomas, especially in the IDH-mutant subtype, creates an environment with low expression of immune checkpoint targets, which provides one possible reason for the resistance to immune checkpoint inhibitors. On the other hand, IDH-mutant tumors are enriched with tumor-resident microglia, which is in contrast to IDH wild-type and brain metastasis that are infiltrated with monocyte-derived macrophages originating from the periphery [8][15][16][8,26,27]. In contrast to brain metastasis, 40% to 70% of the total immune cell populations in glioblastoma are myeloid derived cells, representing the most abundant immune cell type infiltrating these tumors [17][28] that typically have an immune suppressive phenotype.
In addition to the different immune populations that populate different cancer lineages within the CNS, unbiased transcriptional profiling is beginning to reveal distinct subpopulations and states that further increase immunological complexity and heterogeneity even within the same immune cell lineage in the TME. Based on transcriptomics, there may be many different microglia subtypes that demonstrate different functional signatures such as phagocytosis, antigen presentation and lytic functions [18][29]. Although conventional described in the context of M1 (tumor suppressive/immune supportive) or M2 (tumor supportive/immune suppressive) phenotypes, ex vivo data of gliomas have revealed that neither of these were adequate descriptions for tumor associated macrophages (TAMs) [19][30]. With emerging single cell sequencing data, distinct subtypes/subgroups will be emerging that were not evident with bulk profiling initiatives. The innate immune cells in gliomas likely express vastly different molecular phenotypes, transcriptional states and functionalities that are influenced by cancer lineage (gliomas or metastasis), genetic status (IDH-mutation status), and within the TME location (hypoxia) further confounding the heterogeneity of the TME [19][20][30,31] likely even within the same tumor. This area needs further investigation, including functional immune characterization, in order to comprehend the different subtypes of the various tumor associated immune cells and to be fully informed regarding therapeutic selection and optimization.
Additional evidence that the glioma TME is distinct from other cancer lineages is the lack of tumor mutational burden (TMB) being a predictive response biomarker to immune checkpoint inhibitors. McGrail et al. recently showed in an analysis of over 10,000 patients that not all tumors with high-TMB developed effector immune responses to immune checkpoint inhibitors therapy [21][32]. Specifically, in breast, prostate, and glioma cancer patients there appears to be no correlation between CD8 T cell levels and neoantigen load, with overall survival [22][33]. In another analysis of recurrent glioblastoma patients, a very low-TMB state was associated with better response to immune checkpoint inhibition or polio virotherapy [23][34]. The distinction may reside in the mutational composition found in glioma not being particularly immunogenic or alternatively that the mutagenic antigens are eliminated potentially due to better immune surveillance in the patients with lower TMB, resulting in better immune recognition and elimination of mutated sub-clones before the initiation of immunotherapy. Such immune environment would keep the overall TMB low and would explain the otherwise apparent discordance between studies of patients with constitutional DNA mismatch repair syndromes that show responses to immunotherapy. Other investigators have shown that specific mutations like PTEN, are associated with worse outcomes to immune checkpoint inhibitors and one could consider removing these subjects from use of immune checkpoint inhibitors [24][35], rather than enrichment selection based on TMB. In contrast, in recurrent glioblastoma patients that respond to PD-1 blockade, there is enriched expression of BRAF and PTPN11 activation mutations [24][25][35,36]. Arietta et al. showed that phosphorylated ERK1/2 (p-ERK1/2) proteins are predictors of response in recurrent glioblastoma patients to PD-1 inhibition even in absence of BRAF and PTPN11 expression [26][37]. Given the complexity of the steps needed to mount an effective anti-tumor immune response, it is likely that prediction of responsiveness to immunotherapies will not be predicated on a single marker. A systems biology approach possibly using different analysis platforms, through genome sequencing, emerging current single cell analysis, immune landscape observation and functional analysis may need to be consolidated to ultimately predict responses and ultimately understand this unique microenvironment.
3. Differential T Cell Deactivation and Suppression in CNS Cancer Lineages
Multiple immune suppressive mechanisms and pathways are utilized by the tumor cells and their environment to inhibit anti-tumor eradication. T cells are usually the effectors of the cytotoxic anti-tumor immune response in many cancers and if found in the glioblastoma TME, they are under a dysfunctional state and exhausted. Exhaustion occurs after repeated antigen exposure that leads to the expression of multiple immune checkpoints on the T cell surface [27][93]. Brain metastases are more enriched with TILs relative to glioblastoma that have abundant tumor-associated myeloid cells (TAMs) [8][16][8,27]. As such, engagement of immune checkpoints and modulation is more available for targeting in brain metastasis relative to glioblastoma. Notably, the expression of TIM-3 and LAG-3 immune checkpoints is relatively low in gliomas [11], and as such these therapeutic targets are unlikely to be beneficial for the treatment of patients with glioma for most patients and especially as a monotherapy. Both cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed death 1 (PD-1), that are induced in activated T cells providing inhibitory signals by binding to their ligands expressed on the surface of antigen presenting cells or tumor cells ( Figure 1 ), are more frequently expressed. Multiple clinical trials have shown that brain metastases are more responsive to immune checkpoint inhibitors therapies such anti-PD-1 and anti-CTLA-4 [6][28][29][6,94,95] relative to glioblastoma [30][31][96,97]. Other immune checkpoints, like TIGIT, were identified to be highly expressed on T cells, especially cytotoxic CD8 + tumor infiltrating T cells in murine models of glioma [32][19]. More recently, through an analysis of glioma infiltrating T cells, Mathewson et al. found that these cells express natural killer (NK) genes. They demonstrated an enhanced anti-tumor effect of these T cells through the direct blockade of CD161, a NK cell receptor expressed on the T cells surface, or by the inactivation of its respective NK gene KLRB1, highlighting the role of CD161 and potential therapeutic intervention [33][98]. The association of all these immunosuppressive markers on T cells surface induce a state of reduced responsiveness to immune stimulations and thus inhibited effector immune response.
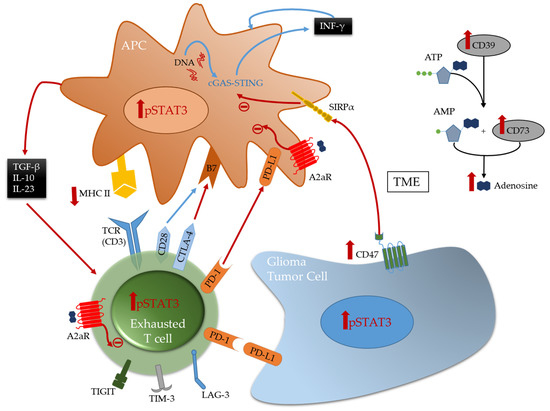
A frequently expressed immune suppressive pathway in glioblastoma is the A2aR-adenosine pathway [34][35][99,100]. Related to this pathway is CD73, an enzyme shown to be preferentially expressed by tumor cells in glioma patients [10] and upregulated in immune cells [11], especially in IDH1-mutant gliomas. This ectonucleotidase is responsible for the activation of adenosine that binds to the A2a receptor on both T cells and myeloid cells thereby triggering immunosuppression [34][99] ( Figure 1 ). Therefore, this pathway is a potential therapeutic target. However, only limited therapeutic benefits were shown by administration of adenosine receptor inhibitors in multiple murine models of gliomas, including those expressing CD73 even in combination with other inhibitors [11]. This highlights the challenges in approaches aimed to reverse T cell exhaustion induced in glioma TME [32][36][19,38].
4. Emerging Strategies for Modulation of Immunosuppressive Tumor Associated Macrophages
TAMs, the most abundant immune cells present in the TME, express a wide variety of immunosuppressive phenotypes [37][101]. Gliomas express CD47 which blocks phagocytosis thereby evading immune recognition and eradication [38][39][102,103]. CD47 blockade in several murine models [38][40][102,104] has shown therapeutic benefit by freeing the innate immune system to activate APCs thereby leading to effector CD8 + effector T cells mediated immune responses [41][42][105,106]. However, monotherapy leads to treatment resistance [42][43][44][106,107,108]. Therefore, combination therapies can represent a solution for better induced anti-tumor responses and outcomes. A recent study demonstrated that combining anti-CD47 targeted therapy with temozolomide creates a pro-phagocytosis effect and induces antigen presentation by activating and upregulating the cGAS-STING pathway in APCs, thus leading to an effective adaptive immune response [44][108].
Stimulator of interferon genes (STING) is an important component of the innate immune response to pathogenic DNA. It is a widely expressed sensor of cellular stress, activated by cytosolic cyclic dinucleotides, which may be released by bacteria or created through cytosolic self- or viral-DNA interaction with cyclic GMP-AMP synthase (cGAS) [45][46][109,110]. STING agonists can induce T cell infiltration into tumors known to be devoid of such immune composition and in tumors enriched with myeloid immune cells through pro-inflammatory activation resulting in marked in vivo therapeutic activity [47][48][111,112]. This pathway bridges the innate and adaptive immune systems both by triggering interferon (IFN) release and through activation of myeloid cells ( Figure 1 ). Distinct from most other innate immune agonists, STING activation can re-educate tumor-supportive immunosuppressive macrophages toward a pro-inflammatory phenotype and can reverse the suppressive phenotype of myeloid-derived suppressor cells (MDSCs) [49][50][113,114]. STING agonists have demonstrated radiographic responses in canines with high-grade gliomas [51][115]. In summary, STING agonists may be a compelling therapeutic strategy for gliomas because they: (1) can simulate a foreign body reaction, thus providing a “target”; (2) induce IFN, thereby providing potent T cell effector action; (3) induce chemokine production and thus T cell trafficking to the tumor; and they are (4) easy to synthesize.
The reciprocal immune modulatory strategy of TAMs is to block a key deactivating pathway the signal transducer and activator of transcription 3 (STAT3). STAT3 is an important mechanism of suppression of both innate and adaptive components of the immune system ( Figure 2 ). STAT3 expression shifts the TAMs to an immunosuppressive phenotype secreting immunomodulatory suppressive factors like IL-10 and TGF-β, and impairing phagocytosis and antigen presentation [52][53][54][116,117,118]. Similarly, STAT3 impairs maturation and antigen presentation by dendritic cells (DCs) preventing T cell activation and proliferation [55][56][119,120]. The inhibition of STAT3 can reactivate the immune system in the TME by promoting infiltrating DCs maturation, increasing expression of the co-stimulatory molecules (CD80/CD86) necessary for T cell activation, and decreasing the number of myeloid derived suppressor cells (MDSCs) in the immune microenvironment [57][58][59][121,122,123]. Additionally, STAT3 is a key inducer of immune suppressive cytokines ( IL-10, IL-4, IL-6 and TGF-β), maintains immunosuppressive cell cross-talk [60][61][62][63][64][124,125,126,127,128], increases tumor infiltration by MDSCs, and induces T cell arrest and apoptosis [65][66][67][68][129,130,131,132]. Through MDSCs secretion of INF-α and other mechanisms, STAT3 upregulates the expression of inhibitory immune checkpoints like PD-L1 on the surface of TAMs and tumor-infiltrating DCs [62][66][126,130]. Furthermore, STAT3 signaling is correlated with a decrease in effector T cells infiltration of the tumor and prevents CD8 + T cell activation by increasing the secretion of INF-γ [69][133]. STAT3 activation correlates with the activation and upregulation of FOXP3 in T cells and is a key inducer of immunosuppressive Tregs infiltration of the TME [70][71][72][73][134,135,136,137]. Several studies, in mice models of glioma and on patient-extracted glioma tumor cells, have demonstrated the benefits of inhibiting STAT3 that enhances the anti-tumor immune response by improving T cell, DCs and NK activation in the TME [55][74][119,138].
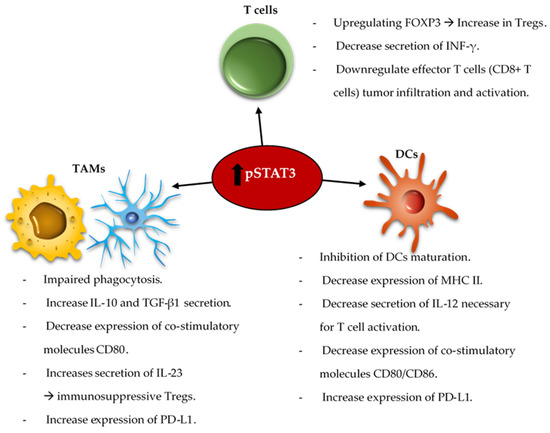
The combination of the standard of care radiation therapy with a STAT3 inhibitor, showed improvement in the overall survival of mice harboring intracranial gliomas [75][139]. This was shown to be immunologically mediated with reprogramming of the DCs in the TME. STAT3 signaling is involved in a variety of intrinsic and acquired resistance mechanisms to administered anti-tumor therapies such as temozolomide [76][77][78][140,141,142], radiation [79][80][81][82][83][84][85][143,144,145,146,147,148,149], and targeted therapies [86][87][88][150,151,152]. As such, some studies demonstrated that the combination of anti-VEGF (Cediranib) and STAT3 inhibitors (AZD1480) for the treatment of glioblastoma in a mice model led to a decrease in the tumor volume and angiogenesis [89][90][153,154]. The co-inhibition of STAT3 and MET induce glioma tumor cells destruction by reactivating apoptosis mechanisms [91][155]. As such, an area of future investigation is combinatorial immune modulatory or chemotherapeutic strategies with STAT3 inhibition.