 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jessica Burket | + 3119 word(s) | 3119 | 2021-08-26 11:23:22 | | | |
| 2 | Amina Yu | Meta information modification | 3119 | 2021-09-10 09:39:53 | | |
Video Upload Options
Data suggest that pathological disturbance of the population of fast-spiking, parvalbumin-expressing GABAergic inhibitory interneurons occur in at least some clinical presentations, which leads to disruption of the synchronous oscillatory output of assemblies of pyramidal neurons. Increased expression of the GluN2A NMDA receptor subunit on parvalbumin-expressing interneurons is linked to functional maturation of both these neurons and the perineuronal nets that surround them. Disruption of GluN2A expression shows increased susceptibility to oxidative stress, reflected in redox dysregulation and delayed maturation of PNNs.
1. Introduction
There are many proteins participating in binding, transport, and distribution of the redox reactive metals iron and copper throughout the body and across membranes, including across neuronal cell membranes and those of specialized organelles within cells. These metal cations are also crucial to the catalytic function of many enzymes and mitochondrial respiration. For example, synaptic release of free copper also acts locally to modulate neurotransmission [1]. Within the developing brain, iron is critical for hippocampal development and the differentiation of parvalbumin-expressing neurons whose soma and dendrites are surrounded by perineuronal nets (PNNs), a specialized elaboration of the extracellular matrix [2][3][4]. Free concentrations of the redox reactive metals iron and copper are highly regulated because of their abilities to catalyze generation of toxic free radical species. The complex architecture of PNNs bind metal cations, which serves a protective function against oxidative damage, especially oxidative damage of the fast-spiking neurons with their dense concentration of rapidly respiring mitochondria [5][6][7][8]. It is conceivable that the binding of these metal redox reactive cations not only contributes to maintenance of their safe local concentrations, but also serves as a reservoir that can “buffer” against second-to-second fluctuations in their concentrations and availability [4].
Histofluorescent-staining of PNNs with specific plant lectins is a useful morphologic approach to identify subpopulations of fast-spiking, parvalbumin-expressing (FSPV+) GABAergic interneurons, as well as populations of pyramidal neurons. Within the cerebral cortex and hippocampus, fast-spiking, PV-expressing GABAergic interneurons are the most populous neurons enwrapped by PNNs, which is consistent with an important role of the PNN in maintaining and protecting energy-efficient and rapid information transfer. These interneurons regulate the synchronous oscillatory output of assemblies of pyramidal output neurons that are computed “upward” into gamma-band synchrony of 30–80 Hz [4][9][10][11]. Gamma-band synchrony across cortical areas facilitates functional connectivity between them and is necessary for performance of many higher cognitive functions, such as working memory, response selection, object representation, attention and sensorimotor integration, among other functions [4][9][10][12][13][14]. Disturbance of these fast-spiking, parvalbumin-expressing neurons leads to disruption of the synchronous oscillatory output of assemblies of pyramidal neurons that ultimately impact functional connectivity and higher cognitive functions ( Figure 1 ).
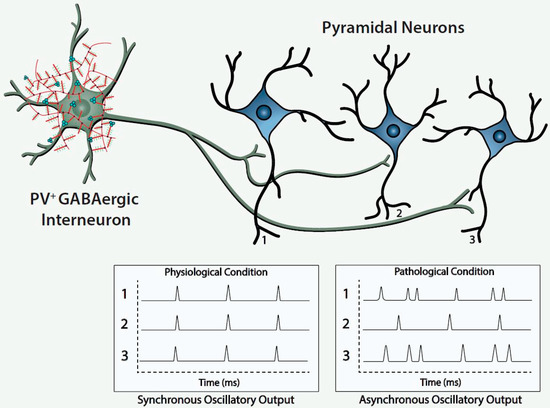
The figure depicts a fast-spiking parvalbumin-expressing GABAergic projection onto an assembly of neocortical pyramidal output neurons. The inhibitory input of the GABAergic projection synchronizes the oscillatory output of the assembled pyramidal neurons, which, in turn, is computed upward into rhythms that may mediate functional connectivity across disparate regions of the cerebral cortex. Conceivably, abnormalities of the handling of redox-reactive metals by the PNNs surrounding the neurons in this circuit would lead to impairment of the fast-spiking capability of the GABAergic projection and/or oxidative damage to the GABAergic projection or pyramidal neurons comprising this neocortical circuit. This pathological disturbance is depicted in the figure by the asynchronous firing of the pyramidal neurons. Ultimately, disruption of fast-spiking and/or oxidative damage would lead to dysrhythmias that impair functional connectivity.
From a neurodevelopmental perspective, morphologically mature PNNs are implicated in the closing of critical periods, which may involve actual obstruction to the entry of neuritic extensions. Thus, therapeutic disruption of mature PNNs is proposed as a mechanism for promoting plasticity in some neurodevelopmental disorder [4]. Neurobiological interest in numerous functional roles of the PNNs themselves continues to grow and the possible contributions of neocortical circuits, comprised of projections of fast-spiking, parvalbumin-expressing GABAergic inhibitory neurons to assemblies of pyramidal neurons, to the pathogenesis of schizophrenia and autism spectrum disorder [3][4][15][16]. Functionally, the PNNs are implicated in the stabilization of synapses and consolidation of long-term memory [4][17][18]. The maturation of the PNN is also implicated in the mechanism of closing critical periods of neural circuit development, creating a physical barrier, thereby reducing neural plasticity [6][14][19]. Although somewhat paradoxical and, perhaps, counter-intuitive, impeding plasticity may assure energy-efficient and more rapid information transfer within dedicated circuits, reflecting selection of shortest path lengths within circuits that are “quiet” and shielded from interference by extraneous “noise”. The current review seeks to provide a brief overview of PNN structure and function and explore a new and emerging area of interest related to the regulation of local concentrations of critical redox metal ions and possible functional consequences and therapeutic potential of their interaction and regulation with PNNs.
2. Architecture and Function of Perineuronal Nets and their Structural Components
The profile of “Selective Neuronal Vulnerability” of different neurons and brain areas to neurofibrillary degeneration is used in neuropathological staging of Alzheimer’s disease (AD) [20]. PNNs bind and scavenge ‘redox active iron”, which is their proposed mechanism of protection against oxidative damage. Consistent with this proposed mechanism of protection, accumulation of lipofuscin within neurons of patients with AD, an intralysosomal pigment generated by iron-catalyzed oxidation, is reduced in those neurons surrounded by PNNs, compared to neurons not surrounded by PNNs. Interestingly, cortical and subcortical neurons enwrapped by PNNs in AD brain are protected against neurofibrillary degeneration. This was demonstrated in a histochemical study that sought to confirm that PNNs are protective against iron-induced oxidative damage using 3-month-old knockout mice with deletions of genes encoding critical PNN structural components to identify if these selectively knocked-out components were necessary for, or contributed to, antioxidant effects [20].
Importantly, there is a dynamic remodeling of GAGs and CSPGs mediated by proteases, glycosidases and sulfatases, whose activities are modulated in response to physiological and pathological processes [14][21]. Sulfation can occur at the C4 and C6 positions of N-acetylgalactosamine and the C2 position of D-glucuronic acid; again, the patterns of sulfation are neither invariant nor random and can have consequences for the free extracellular calcium ion concentration and transmembrane potential. Purified solutions of chondroitin 4 sulfate (CS4) and chondroitin 6 sulfate (CS6) were used to measure their calcium ion binding properties [21]. Very briefly, the construction of chelation curves to measure the binding capabilities of free calcium ions involved incubating purified solutions of CS4 and CS6 with solutions of free calcium. Hyaluronan lacks sulfation and, thus, was shown to have limited calcium ion binding capability. Bound calcium ions were dissociated from the purified chondroitin sulfate preparations, and the amount of dissociated bound calcium ion was measured. The functional significance of differences in the binding capabilities of CS4 and CS6 with calcium ions was studied with electrophysiological recordings from light-adapted principal rods of single cells; photoreceptor suspensions were prepared from retina of Xenopus laevis .
As discussed, the GAG components of the PNNs are responsible for dense negative charges in the microenvironment of the subset of neurons they ensheath [22]. These fixed anionic binding sites influence diffusion properties of charged diffusing particles and acts as an immobilized ion exchanger, which partitions ions according to their charge and Donnan equilibrium ( Figure 2 , lower left panel). Using a novel approach (i.e., “high resolution nuclear microscopy with focused proton-beam microprobes in combination with an ionic metallic probe”), fixed negative charge density in the neuronal microenvironment were visualized and quantified in brain slices; also, the methodology enabled measurement of bound cationic probes, such as Fe 3+ ions, within PNNs [22]. Relative amounts of iron atoms per sulfur atoms in PNNs varied according to brain region, ranging from a high of ~3.8 in the subiculum to a low of ~1.3 in the red nucleus. This binding reflects the relative amount of iron ions bound to the chondroitin sulfate disaccharide units because iron labeling disappeared when brain sections were treated with chondroitinase or hyaluronidase, which removes GAG side chains. Moreover, the specialized PNNs accumulated two to three times more Fe 3+ than ECM in general. The anionic charge density of the PNNs afforded estimates of the maximal binding of Fe 3+ , which were in the 117 to 180 mM range (which are much higher estimates than previously suspected). Diffusion properties in regions enriched with PNNs are anisotropic, reflecting their interference with free diffusion.
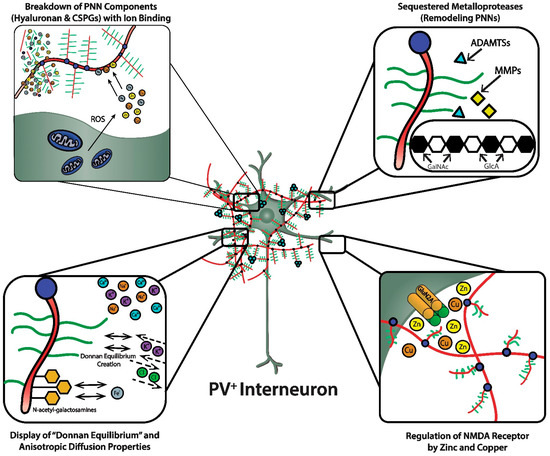
A complex enzymology is involved in the dynamic remodeling of PNNs that almost certainly affects learning, memory and efficiency of information transfer within neural circuits; moreover, their remodeling may be part of pathogenic mechanisms in a variety of CNS disorders [3][14][15][23]. In fact, there is already therapeutic interest in selective inhibitors of several of these remodeling enzymes. In addition to functions attributed to their physicochemical properties, such as ion sorting, anionic shielding, and the impact of the Donnan equilibrium on rapid adjustments of membrane potential, the three-dimensional scaffolding structure of PNNs in particular, and the ECM in general, affects signal transduction through their interactions with molecules on the cell surface, including integrins and cadherins, brain development through regulation of cell migration, and both normal and pathological aging [14].
3. Copper Homeostasis, Brain Development, and Functional Consequences of Binding to PNNs
Copper ions play familiar biological roles in terms of mitochondrial respiration and ATP production and protection against oxidative stress and damage; moreover, unbound redox reactive copper is itself a mediator of oxidative stress and damage. The transition of unbound copper between its reduced Cu + and oxidized Cu 2+ states can lead to production of the highly reactive hydroxyl radical (OH) − through the reaction of the superoxide anion (O 2− ) and hydrogen peroxide (H 2O 2), which can attack proteins, lipids and nucleic acids [1]. Furthermore, unbound copper can play toxic roles by catalyzing interactions of nitric oxide (NO), a gaseous retrograde novel neurotransmitter that regulates receptor-mediated changes in intraneuronal levels of cGMP, with O 2− or endogenous thiols producing the reactive nitrogen species (RNS) peroxynitrite (CNOO − ) or S-nitroso-thiols, respectively [1]. Both reactive oxygen species (ROS) and RNS are implicated in the pathogenesis of neurodegenerative diseases, such as Parkinson’s disease (PD), Alzheimer’s disease (AD), and amyotrophic lateral sclerosis (ALS). Thus, the heightened importance of handling the essential copper ion, especially in anatomic areas of brain whose function depends on highly active mitochondrial respiration, such as fast-spiking, PV-expressing GABAergic interneurons in neocortex and hippocampus [4][10][24].
The essential nature of copper to neurologic function and the pathological consequences of disturbances of copper levels, distribution and/or binding are highlighted by both Menkes and Wilson’s diseases, which are characterized by mutations in the copper transport genes ATP7A and ATB7B , respectively [25]. On the apical side of the endothelial cell, ATP7B, a P-type ATPase, transports excess free copper out of the endothelial cell going from the direction of the brain to the general circulation. Wilson’s disease is due to an autosomal homozygous recessive mutation of ATB7B located on chromosome 13 [26]. On the basolateral side of the BBB’s endothelial cells, ATP7A, a P-type ATPase that shares about 55% homology with ATP7B, works to transport free copper into the brain parenchyma. ATP7A resides within the membrane of the trans -Golgi network (TGN), where it transports free copper from the cytosol into the lumen of this secretory network for incorporation into cuproenzymes. However, in the event of copper overload, ATP7A is translocated from the TGN in large free copper-containing cytosolic vesicles for incorporation into the plasma membrane and release of excess free copper. In Menkes disease, an X-linked disorder associated with almost 300 loss of function mutations of the gene encoding ATP7A, whose phenotype includes systemic copper deficiency and pleiotropic CNS manifestations, there is cerebrovascular copper accumulation and low brain copper levels [1][25]. Menkes disease highlights the importance of highly regulated handling and transport of copper for CNS function and development; most notably, in Menkes disease, genetic defects in a P-type ATPase causes failures to transport copper across the placenta, GI tract, and BBB. Menkes disease leads to death within the first decade, and demyelination and neurodegeneration are found in cerebral cortex, hippocampus, thalamus and cerebellum [27]. Similarly, acquired copper deficiencies due to diverse etiologies (e.g., excessive zinc consumption, gastrectomy and idiopathic malabsorption) are associated with neurological presentations, primarily affecting the peripheral nervous system. In terms of the blood/CSF barrier, Ctr1 transports free copper from the CSF into the epithelial cell, whereas ATP7A transports free copper into the CSF. ATP7B is located on the other pole of the blood/CSF barrier to transport free copper into blood.
Because of the potential toxicity of free copper, there are copper chaperone proteins (referred to as metallochaperones) that direct copper to specific targets and pathways through protein-protein interactions (e.g., inserting copper into the active sites of cuproenzymes and transport proteins) [27]. Examples of chaperones include atox1, which transfers copper to P-type ATPases, and cox17, which delivers copper to the mitochondria where it is necessary for assembly of cytochrome C oxidase. P-Type ATPases are not only involved in the directional transport of copper into and out of endothelial and epithelial cells of the BBB and blood/CSF barrier, but are also necessary for compartmentalization of copper within the cell (e.g., transport into the Golgi apparatus for incorporation into secretory proteins and creation of a free pool of releasable copper).
Once copper has entered the cell, glutathione (GSH) has roles in sequestering copper and, thereby, helping to prevent its toxic accumulation, and its intracellular transport between Ctr1 and copper chaperone proteins [26]. GSH, a soluble tripeptide composed of glutamate, cysteine and glycine, is an antioxidant that acts by itself or as an enzyme cofactor of glutathione peroxidases and glutathione S-transferases to protect against ROS and RNS. In addition to its familiar role to protect against oxidative damage, disturbances of the tightly regulated levels of GSH are associated with pathological disruption of both intracellular copper transport and maintenance of safe nontoxic levels of free copper [26]. For example, an increased ratio of the reduced GSH to glutathione disulfide, the oxidized product resulting from the reaction of GSH with radical species, is observed in differentiated cultured neurons compared to non-differentiated neurons. This equilibrium shift to GSH in differentiated motor neurons is associated with increased ATP7A-dependent copper efflux via the secretory pathway. The bivalent oxidized state of copper interferes with protein degradation by the ubiquitin proteasome system by promoting aggregation and self-oligomerization of ubiquitin, which reduces the pool of free ubiquitin and degradation of misfolded proteins [26]. Moreover, genetic manipulation using RNAi to knockdown GSH biosynthesis in Drosophila caused neuronal defects and embryonic lethality that was attenuated with copper supplementation [26]. Conceivably, pathogenic mechanisms of at least some neurodegenerative diseases could include reductions of cytosolic levels of GSH associated with abnormalities in transport and intraneuronal distribution of copper, and increased intraneuronal levels of Cu 2+ , which, in turn, could result in abnormal protein aggregation, impaired protein degradation, and oxidative damage [26].
4. NMDA Receptor and PNN Function: Effects of Oxidative Stress and Therapeutic Potential
Relative expression of the heterotetrameric GluN2A ionotropic NMDA receptor containing two obligatory GluN1 subunits and two variable GluN2A subunits, compared to the heterotetrameric GluN2B receptor, increases in postnatal life [8][28]. The increased expression of GluN2A receptors on PV-expressing interneurons is linked to functional maturation of these neurons and the PNNs that surround them and cortical network refinement. Mice lacking expression of GluN2A receptors have increased susceptibility to oxidative stress, which is reflected in redox dysregulation and delayed maturation of PNNs [8]. Increased susceptibility to sub-threshold oxidative stress and redox dysregulation, as well as delayed maturation of PV-expressing neurons and their associated PNNs were studied in the anterior cingulate cortex (ACC) of knockout (KO) mice lacking GluN2A expression (GRIN2A KO mice) not exposed and exposed to GBR12909 (GBR; 10 mg/kg, i.p.), a dopamine transporter inhibitor and cause of mild oxidative stress, over postnatal days (PDs) 10 to 20 [8]. At PD20, the intensity of both PV-immunostaining in neuronal processes and histofluorescent-staining of PNNs with Wisteria floribunda agglutinin (WFA) in ACC of GRIN2A KO mice was lower, while the number of PV-expressing cells did not differ between GRIN2A KO mice and wild-type mice. At both PD20 and PD60, a significantly lower percentage of PV-expressing cells in the ACC of GRIN2A KO mice were wrapped with PNNs fluorescently-tagged with WFA than PV-expressing cells in the ACC of wild-type mice.
The GRIN2A KO mice subjected to 10-days of GBR12909-induced mild oxidative stress, which generates ROS, resulted in significantly more intensely-stained 8-oxo-2′-deoxyguanosine (8-oxo-dG) immunoreactivity, a marker of oxidative damage, compared to PBS- and GBR-treated wild-type mice. This mild GBR-induced oxidative stress in the GRIN2A KO mice during PDs 10–20 led to reductions of PV-expressing neurons, PV expression, and less intense WFA-fluorescently-stained PNNs; comparisons were made to PBS- and GBR-treated wild-type mice [8]. The less intense WFA-fluorescently-stained PNNs of the mild GBR-induced oxidatively stressed GRIN2A KO mice may reflect their “chemical” immaturity. Functionally, the chemical maturity of PNNs, which is also reflected in the sulfation patterns of their chondroitin sulfate proteoglycans, is necessary for their sequestration of semaphorin 3A, a chemorepulsive axon guidance protein, and Otx2 (orthodenticle homeobox 2), an important homeobox transcription factor [4][22][29]. The severity of these changes in the GBR-treated GRIN2A KO offspring were attenuated by adding N-acetylcysteine, which possesses antioxidant properties, to the drinking water (2.4 g/L) of the lactating mothers from PDs7 to 20 [8].
The heterotetrameric GluN2A receptor is a hypothesized therapeutic target for maintaining synchronous oscillatory output of assemblies of neocortical pyramidal neurons [4][28]. The maturity of PNNs is necessary for differentiation and PV expression of GABAergic interneurons that synchronize this oscillatory output [4][10]. In addition to their roles in refining neural circuitry and closing critical periods, based on their physicochemical properties, PNNs play important roles in protecting against oxidative damage and providing a reservoir of metal and other cations that are critical for NMDA receptor function in the local microenvironment of neocortical neuronal circuits ( Figure 2 , lower right panel). For example, NMDA receptor function depends on membrane potential-dependent binding of the hydrated magnesium ion to a channel domain of this ionotropic receptor, and binding of zinc contributes to regulation of glutamate-gated channel opening [28][30].
The zinc bivalent cation bound to hyaluronic acid can act as an antioxidant and protect hyaluronic acid against oxidative damage by displacing iron ions from their binding sites [31]. Zn 2+ showed a dose-dependent protective effect against the superoxide radical-induced damage of hyaluronic acid and a “moderate” protective effect against this damage by peroxynitrite. These protective “scavenger” effects of Zn 2+ were related to its interaction with hyaluronic acid [31]. Importantly, these data show that zinc binds to the hyaluronic acid backbone of the PNN, consistent with the possibility that this binding by the PNN may be relevant to maintenance and fluctuations of local zinc concentrations in the microenvironments of specific neuronal populations and gating of ionotropic NMDA receptors by glutamate and glycine/D-serine.
References
- D’Ambrosi, N.; Rossi, L. Copper at Synapse: Release, Binding and Modulation of Neurotransmission. Neurochem. Int. 2015, 90, 36–45.
- Callahan, L.S.N.; Thibert, K.A.; Wobken, J.D.; Georgieff, M.K. Early-Life Iron Deficiency Anemia Alters the Development and Long-Term Expression of Parvalbumin and Perineuronal Nets in the Rat Hippocampus. Dev. Neurosci. 2013, 35, 427–436.
- Balmer, T.S. Perineuronal Nets Enhance the Excitability of Fast-Spiking Neurons. Eneuro 2016, 3.
- Burket, J.A.; Urbano, M.R.; Deutsch, S.I. Sugarcoated Perineuronal Nets Regulate “GABAergic” Transmission: Bittersweet Hypothesis in Autism Spectrum Disorder. Clin. Neuropharmacol. 2017, 40, 120–130.
- Cabungcal, J.-H.; Steullet, P.; Morishita, H.; Kraftsik, R.; Cuenod, M.; Hensch, T.K.; Do, K.Q. Perineuronal Nets Protect Fast-Spiking Interneurons against Oxidative Stress. Proc. Natl. Acad. Sci. USA 2013, 110, 9130–9135.
- Do, K.Q.; Cuenod, M.; Hensch, T.K. Targeting Oxidative Stress and Aberrant Critical Period Plasticity in the Developmental Trajectory to Schizophrenia. Schizophr. Bull. 2015, 41, 835–846.
- Steullet, P.; Cabungcal, J.-H.; Coyle, J.; Didriksen, M.; Gill, K.; Grace, A.A.; Hensch, T.K.; LaMantia, A.-S.; Lindemann, L.; Maynard, T.M.; et al. Oxidative Stress-Driven Parvalbumin Interneuron Impairment as a Common Mechanism in Models of Schizophrenia. Mol. Psychiatry 2017, 22, 936–943.
- Cardis, R.; Cabungcal, J.-H.; Dwir, D.; Do, K.Q.; Steullet, P. A Lack of GluN2A-Containing NMDA Receptors Confers a Vulnerability to Redox Dysregulation: Consequences on Parvalbumin Interneurons, and Their Perineuronal Nets. Neurobiol. Dis. 2018, 109, 64–75.
- Bartos, M.; Vida, I.; Jonas, P. Synaptic Mechanisms of Synchronized Gamma Oscillations in Inhibitory Interneuron Networks. Nat. Rev. Neurosci. 2007, 8, 45–56.
- Deutsch, S.I.; Rosse, R.B.; Schwartz, B.L.; Mastropaolo, J.; Burket, J.A.; Weizman, A. Regulation of Intermittent Oscillatory Activity of Pyramidal Cell Neurons by GABA Inhibitory Interneurons Is Impaired in Schizophrenia: Rationale for Pharmacotherapeutic GABAergic Interventions. Isr. J. Psychiatry Relat. Sci. 2010, 47, 17–26.
- Murthy, S.; Kane, G.A.; Katchur, N.J.; Lara Mejia, P.S.; Obiofuma, G.; Buschman, T.J.; McEwen, B.S.; Gould, E. Perineuronal Nets, Inhibitory Interneurons, and Anxiety-Related Ventral Hippocampal Neuronal Oscillations Are Altered by Early Life Adversity. Biol. Psychiatry 2019, 85, 1011–1020.
- Carlén, M.; Meletis, K.; Siegle, J.H.; Cardin, J.A.; Futai, K.; Vierling-Claassen, D.; Rühlmann, C.; Jones, S.R.; Deisseroth, K.; Sheng, M.; et al. A Critical Role for NMDA Receptors in Parvalbumin Interneurons for Gamma Rhythm Induction and Behavior. Mol. Psychiatry 2012, 17, 537–548.
- Gonzalez-Burgos, G.; Cho, R.Y.; Lewis, D.A. Alterations in Cortical Network Oscillations and Parvalbumin Neurons in Schizophrenia. Biol. Psychiatry 2015, 77, 1031–1040.
- Krishnaswamy, V.R.; Benbenishty, A.; Blinder, P.; Sagi, I. Demystifying the Extracellular Matrix and Its Proteolytic Remodeling in the Brain: Structural and Functional Insights. Cell Mol. Life Sci. 2019, 76, 3229–3248.
- Bitanihirwe, B.K.Y.; Woo, T.-U.W. Perineuronal Nets and Schizophrenia: The Importance of Neuronal Coatings. Neurosci. Biobehav Rev. 2014, 45, 85–99.
- Berretta, S.; Pantazopoulos, H.; Markota, M.; Brown, C.; Batzianouli, E.T. Losing the Sugar Coating: Potential Impact of Perineuronal Net Abnormalities on Interneurons in Schizophrenia. Schizophr. Res. 2015, 167, 18–27.
- Gogolla, N.; Caroni, P.; Lüthi, A.; Herry, C. Perineuronal Nets Protect Fear Memories from Erasure. Science 2009, 325, 1258–1261.
- Thompson, E.H.; Lensjø, K.K.; Wigestrand, M.B.; Malthe-Sørenssen, A.; Hafting, T.; Fyhn, M. Removal of Perineuronal Nets Disrupts Recall of a Remote Fear Memory. Proc. Natl. Acad. Sci. USA 2018, 115, 607–612.
- Pantazopoulos, H.; Berretta, S. In Sickness and in Health: Perineuronal Nets and Synaptic Plasticity in Psychiatric Disorders. Neural Plasticity 2015, 2016, e9847696.
- Suttkus, A.; Rohn, S.; Weigel, S.; Glöckner, P.; Arendt, T.; Morawski, M. Aggrecan, Link Protein and Tenascin-R Are Essential Components of the Perineuronal Net to Protect Neurons against Iron-Induced Oxidative Stress. Cell Death Dis. 2014, 5, e1119.
- Vigetti, D.; Andrini, O.; Clerici, M.; Negrini, D.; Passi, A.; Moriondo, A. Chondroitin Sulfates Act as Extracellular Gating Modifiers on Voltage-Dependent Ion Channels. Cell Physiol. Biochem. 2008, 22, 137–146.
- Morawski, M.; Reinert, T.; Meyer-Klaucke, W.; Wagner, F.E.; Tröger, W.; Reinert, A.; Jäger, C.; Brückner, G.; Arendt, T. Ion Exchanger in the Brain: Quantitative Analysis of Perineuronally Fixed Anionic Binding Sites Suggests Diffusion Barriers with Ion Sorting Properties. Sci. Rep. 2015, 5, 16471.
- Walker, C.; Mojares, E.; Del Río Hernández, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028.
- Borchard, S.; Bork, F.; Rieder, T.; Eberhagen, C.; Popper, B.; Lichtmannegger, J.; Schmitt, S.; Adamski, J.; Klingenspor, M.; Weiss, K.-H.; et al. The Exceptional Sensitivity of Brain Mitochondria to Copper. Toxicol. Vitro 2018, 51, 11–22.
- Madsen, E.; Gitlin, J.D. Copper and Iron Disorders of the Brain. Annu. Rev. Neurosci 2007, 30, 317–337.
- Greenough, M.A.; Ramírez Munoz, A.; Bush, A.I.; Opazo, C.M. Metallo-Pathways to Alzheimer’s Disease: Lessons from Genetic Disorders of Copper Trafficking. Metallomics 2016, 8, 831–839.
- Schlief, M.L.; Gitlin, J.D. Copper Homeostasis in the CNS: A Novel Link between the NMDA Receptor and Copper Homeostasis in the Hippocampus. Mol. Neurobiol. 2006, 33, 81–90.
- Burket, J.A.; Deutsch, S.I. Metabotropic Functions of the NMDA Receptor and an Evolving Rationale for Exploring NR2A-Selective Positive Allosteric Modulators for the Treatment of Autism Spectrum Disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 90, 142–160.
- de Winter, F.; Kwok, J.C.F.; Fawcett, J.W.; Vo, T.T.; Carulli, D.; Verhaagen, J. The Chemorepulsive Protein Semaphorin 3A and Perineuronal Net-Mediated Plasticity. Neural Plast. 2016, 3679545.
- Deutsch, S.I.; Burket, J.A.; Benson, A.D.; Urbano, M.R. NMDA Agonists for Autism Spectrum Disorders: Progress and Possibilities. Futur. Neurol. 2015, 10, 485–500.
- Balogh, G.T.; Illés, J.; Székely, Z.; Forrai, E.; Gere, A. Effect of Different Metal Ions on the Oxidative Damage and Antioxidant Capacity of Hyaluronic Acid. Arch Biochem. Biophys. 2003, 410, 76–82.

