 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Juan Ling | + 3558 word(s) | 3558 | 2021-08-20 11:53:24 | | | |
| 2 | Beatrix Zheng | + 307 word(s) | 3865 | 2021-08-27 10:26:34 | | | | |
| 3 | Beatrix Zheng | + 307 word(s) | 3865 | 2021-08-27 10:27:41 | | | | |
| 4 | Beatrix Zheng | + 307 word(s) | 3865 | 2021-08-27 10:28:49 | | |
Video Upload Options
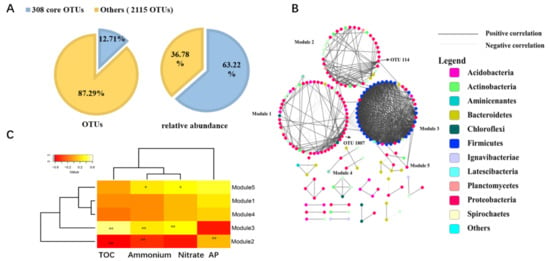
Seagrasses are an important part of the coral reef ecosystem, and their rhizosphere microbes are of great ecological importance. However, variations in diversity, composition, and potential functions of bacterial communities in the seagrass rhizosphere of coral reef ecosystems remain unclear. This study employed the high-throughput sequencing based on 16S rDNA gene sequences and functional annotation of prokaryotic taxa (FAPROTAX) analysis to investigate these variations based on seagrass species and sampling locations, respectively. Results demonstrated that the seagrass rhizosphere microbial community was mainly dominated by phylum Proteobacteria (33.47%), Bacteroidetes (23.33%), and Planctomycetes (12.47%), while functional groups were mainly composed of sulfate respiration (14.09%), respiration of sulfur compounds (14.24%), aerobic chemoheterotrophy (20.87%), and chemoheterotrophy (26.85%). Significant differences were evident in alpha diversity, taxonomical composition and putative functional groups based on seagrass species and sampling locations. Moreover, the core microbial community of all investigated samples was identified, accounting for 63.22% of all obtained sequences. Network analysis indicated that most microbes had a positive correlation (82.41%), and two module hubs (phylum Proteobacteria) were investigated. Furthermore, a significant positive correlation was found between the OTUs numbers obtained and the functional groups assigned for seagrass rhizosphere microbial communities (p < 0.01). Our result would facilitate future investigation of the function of seagrass rhizosphere microbes.
1. Introduction
2. Analysis on Results
2.1. Sampling Locations and Physicochemical Properties of the Seagrass Rhizosphere
| Sample | DYH | SYT | XST | XSH | XSC | XSS | NXH | NXT | |
|---|---|---|---|---|---|---|---|---|---|
| Latitude (° E) | 22.54 | 18.2 | 16.84 | 16.84 | 16.84 | 16.84 | 9.71 | 10.22 | |
| Longtitude (° N) | 114.45 | 119.47 | 112.34 | 112.34 | 112.34 | 112.34 | 114.29 | 114.2 | |
| Water | pH | 8.14 (0) |
8.15 (0.01) |
8.24 (0.01) |
8.24 (0.03) |
8.24 (0.03) |
8.24 (0.01) |
8.15 (0.01) |
8.21 (0.01) |
| Salinity | 28.37 (0.07) |
24.7 (0.3) |
34.52 (0.05) |
34.52 (0.15) |
34.57 (0.08) |
34.52 (0.05) |
34.31 (0) |
34.54 (0.09) | |
| DO (mg/L) | 5.82 (0.05) |
6.7 (0.2) |
9.47 (0.17) |
9.47 (0.51) |
9.64 (0.59) |
9.47 (0.17) |
7.64 (0.03) |
8.36 (0.13) | |
| Nitrate (mg/L) | 0.153 (0.004) | 0.044 (0.002) | 0.049 (0.002) | 0.049 (0.005) | 0.046 (0.001) | 0.049 (0.002) | 0.06 (0.001) |
0.035 (0) |
|
| Nitrite (mg/L) | 0.001 (0) |
0.025 (0.001) | 0.016 (0) |
0.016 (0.001) | 0.016 (0.001) | 0.016 (0) |
0.001 (0) |
0.001 (0) |
|
| Ammonium (mg/L) | 0.069 (0) |
0.121 (0.002) | 0.094 (0.002) | 0.094 (0.005) | 0.091 (0.006) | 0.094 (0.002) | 0.048 (0.001) | 0.032 (0.001) | |
| Phosphate (mg/L) | 0.007 (0.001) | 0.015 (0) |
0.01 (0.001) |
0.01 (0.002) |
0.01 (0.001) |
0.01 (0.001) |
0.006 (0) |
0.006 (0) |
|
| Sediment | Ammonium (mg/kg) | 3.02 (0.01) |
5.97 (0.02) |
4.37 (0.01) |
2.91 (0.03) |
1.45 (0.01) |
4.78 (0.02) |
6.39 (0.03) |
5.48 (0.12) |
| Nitrate (mg/kg) | 22.3 (2.63) |
24.4 (3.68) |
44.0 (2.10) |
62.4 (3.26) |
69.4 (4.06) |
44.0 (2.24) |
32.60 (1.12) |
45.60 (1.56) | |
| AP (mg/kg) | 18.0 (2.01) |
15.0 (1.28) |
14.0 (3.06) |
11.0 (1.85) |
15.0 (2.30) |
15.0 (1.60) |
18 (2.45) |
17 (3.01) |
|
| TOC (%) | 18.4% (1.89) | 32.5% (2.36) |
43.4% (1.03) |
33.2% (2.64) |
40.3% (3.12) | 21.8% (3.56) |
36.80 (2.63) |
42.06 (3.42) |
2.2. Taxonomy, Phylogenetic Diversity, and Composition of Rhizosphere Bacterial Communities of the Seagrass
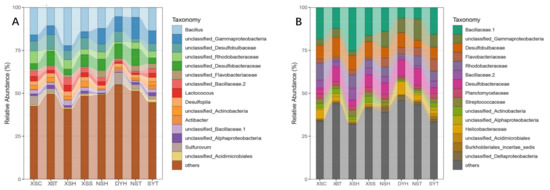
| Microbial Communities |
Microbial Communities |
Phylogenetic Composition (PD) |
Taxonomic Alpha Diversity |
|||
|---|---|---|---|---|---|---|
| Richness | Shannon | Simpson | ||||
| Species | p | p | p | p | ||
| XSC | XST | 0.9420 | 0.6700 | 0.5250 | 0.3240 | |
| XSC | XSH | 0.4550 | 0.3870 | 0.7380 | 0.7380 | |
| XSC | XSS | 0.5250 | 0.6700 | 0.3240 | 0.5250 | |
| XST | XSH | 0.0802 | 0.9690 | 0.0810 | 0.0330 | |
| XST | XSS | 0.2180 | 0.1060 | 0.9870 | 0.9870 | |
| XSS | XSH | 0.0240 | 0.0330 | 0.0330 | 0.0810 | |
| Location | ||||||
| H. ovalis | XSH | DYH | 0.9500 | 0.3700 | 0.0200 | 0.0200 |
| XSH | CGX | 0.2300 | 0.3700 | 0.3700 | 0.3700 | |
| DYH | CGX | 0.1300 | 0.0200 | 0.3700 | 0.3700 | |
| T. hemprichii | ||||||
| XST | SYT | 0.5490 | 0.5500 | 0.3700 | 0.8960 | |
| XST | NXT | 0.5490 | 0.3000 | 0.0370 | 0.0650 | |
| SYT | NXT | 0.0930 | 0.0300 | 0.0200 | 0.1730 | |
| Microbial Community |
Microbial Community |
Delta Unifrac |
P Unifrac |
Delta Bray |
P Bray |
Delta Euclidean |
P Euclidean |
Delta Sorensen |
P Sorensen |
|
|---|---|---|---|---|---|---|---|---|---|---|
| Species | ||||||||||
| XSC | XST | 0.184 | 0.087 | 0.294 | 0.016 | 1.260 | 0.156 | 0.099 | 0.293 | |
| XSC | XSH | 0.177 | 0.084 | 0.308 | 0.109 | 1.590 | 0.294 | 0.152 | 0.282 | |
| XSC | XSS | 0.049 | 0.100 | 0.088 | 0.100 | 1.040 | 0.500 | 0.061 | 0.300 | |
| XST | XSH | 0.219 | 0.025 * | 0.366 | 0.015 * | 1.429 | 0.017 * | 0.136 | 0.124 | |
| XST | XSS | 0.176 | 0.035 * | 0.284 | 0.017 * | 1.073 | 0.009 ** | 0.083 | 0.016 * | |
| XSH | XSS | 0.169 | 0.011 * | 0.299 | 0.012 * | 1.403 | 0.037 * | 0.135 | 0.109 | |
| Location | ||||||||||
| H. ovalis | XSH | NSH | 0.039 | 0.100 | 0.088 | 0.100 | 1.024 | 0.100 | 0.111 | 0.100 |
| XSH | DYH | 0.035 | 0.100 | 0.125 | 0.100 | 1.024 | 0.100 | 0.114 | 0.100 | |
| NSH | DYH | 0.033 | 0.100 | 0.122 | 0.100 | 0.667 | 0.100 | 0.042 | 0.100 | |
| T. hemprichii | XST | NST | 0.070 | 0.100 | 0.110 | 0.100 | 20.104 | 0.100 | 0.130 | 0.100 |
| XST | SYT | 0.064 | 0.100 | 0.112 | 0.100 | 19.395 | 0.100 | 0.132 | 0.100 | |
| NST | SYT | 0.026 | 0.100 | 0.105 | 0.100 | 18.762 | 0.100 | 0.121 | 0.100 |
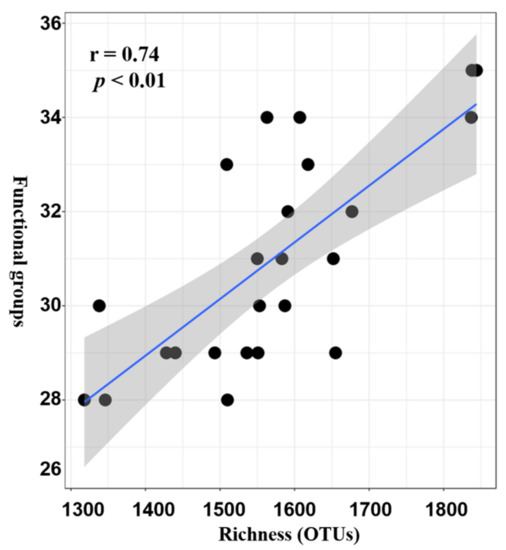
2.3. Potential Functional Roles of Microbial Played in Seagrass Rhizosphere
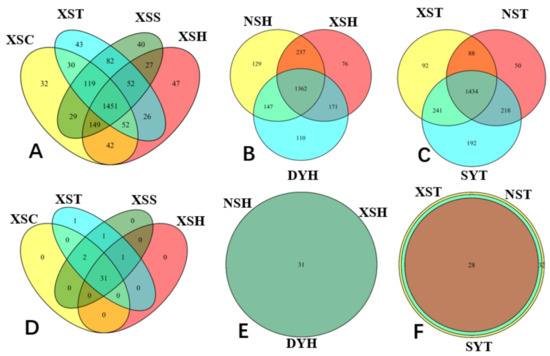
2.4. Venn Diagram Analysis of the Variations in Taxonomy Species and Functional Groups
2.5. Core Microbial Community in Seagrass Microbial Rhizosphere
3. Current Insights
3.1. Variations in the Taxonomical, Phylogenetical Diversity and Composition of Bacterial Communities
3.2. The Functional Structure of Microbial Communities in Seagrass Rhizosphere
3.3. The Core Microbial Community in Seagrass Rhizosphere across the Four Coral Reef Ecosystems
References
- Fraser, M.W.; Gleeson, D.B.; Grierson, P.F.; Laverock, B.; Kendrick, G.A. Metagenomic Evidence of Microbial Community Responsiveness to Phosphorus and Salinity Gradients in Seagrass Sediments. Front. Microbiol. 2018, 9, 1703.
- Hurtado-McCormick, V.; Kahlke, T.; Petrou, K.; Jeffries, T.; Ralph, P.J.; Seymour, J.R. Regional and Microenvironmental Scale Characterization of the Zostera muelleri Seagrass Microbiome. Front. Microbiol. 2019, 10, 1011.
- Conte, C.; Rotini, A.; Manfra, L.; D’Andrea, M.M.; Winters, G.; Migliore, L. The Seagrass Holobiont: What We Know and What We Still Need to Disclose for Its Possible Use as an Ecological Indicator. Water 2021, 13, 406.
- Garcias-Bonet, N.; Eguiluz, V.M.; Diaz-Rua, R.; Duarte, C.M. Host-association as major driver of microbiome structure and composition in Red Sea seagrass ecosystems. Environ. Microbiol. 2021, 23, 2021–2034.
- Ugarelli, K.; Chakrabarti, S.; Laas, P.; Stingl, U. The Seagrass Holobiont and Its Microbiome. Microorganisms 2017, 5, 81.
- Cúcio, C.; Overmars, L.; Engelen, A.H.; Muyzer, G. Metagenomic Analysis Shows the Presence of Bacteria Related to Free-Living Forms of Sulfur-Oxidizing Chemolithoautotrophic Symbionts in the Rhizosphere of the Seagrass Zostera marina. Front. Mar. Sci. 2018, 5, 171.
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 2013, 37, 634–663.
- Cúcio, C.; Engelen, A.H.; Costa, R.; Muyzer, G. Rhizosphere Microbiomes of European Seagrasses Are Selected by the Plant, But Are Not Species Specific. Front. Microbiol. 2016, 7, 440.
- Borum, J.; Pedersen, O.; Greve, T.M.; Frankovich, T.A.; Zieman, J.C.; Fourqurean, J.W.; Madden, C.J. The potential role of plant oxygen and sulphide dynamics in die-off events of the tropical seagrass, Thalassia testudinum. J. Ecol. 2005, 93, 148–158.
- Pedersen, M.F.; Borum, J. Nitrogen Dynamics of Eelgrass Zostera-Marina during a Late Summer Period of High Growth and Low Nutrient Availability. Mar. Ecol. Prog. Ser. 1992, 80, 65–73.
- Agawin, N.S.R.; Ferriol, P.; Sintes, E.; Moya, G. Temporal and spatial variability of in situ nitrogen fixation activities associated with the Mediterranean seagrass Posidonia oceanica meadows. Limnol. Oceanogr. 2017, 62, 2575–2592.
- Pereg, L.L.; Lipkin, Y.; Sar, N. Different Niches of the Halophila-Stipulacea Seagrass Bed Harbor Distinct Populations of Nitrogen-Fixing Bacteria. Mar. Biol. 1994, 119, 327–333.
- Brodersen, K.E.; Siboni, N.; Nielsen, D.A.; Pernice, M.; Ralph, P.J.; Seymour, J.; Kuhl, M. Seagrass rhizosphere microenvironment alters plant-associated microbial community composition. Environ. Microbiol. 2018, 20, 2854–2864.
- Shade, A.; Handelsman, J. Beyond the Venn diagram: The hunt for a core microbiome. Environ. Microbiol. 2012, 14, 4–12.
- Jia, X.; Li, X.D.; Zhao, Y.H.; Wang, L.; Zhang, C.Y. Soil microbial community structure in the rhizosphere of Robinia pseudoacacia L. seedlings exposed to elevated air temperature and cadmium-contaminated soils for 4 years. Sci. Total Environ. 2019, 650, 2355–2363.
- Li, J.; Luo, Z.; Zhang, C.; Qu, X.; Chen, M.; Song, T.; Yuan, J. Seasonal Variation in the Rhizosphere and Non-Rhizosphere Microbial Community Structures and Functions of Camellia yuhsienensis Hu. Microorganisms 2020, 8, 1385.
- Sun, F.; Zhang, X.; Zhang, Q.; Liu, F.; Zhang, J.; Gong, J. Seagrass (Zostera marina) Colonization Promotes the Accumulation of Diazotrophic Bacteria and Alters the Relative Abundances of Specific Bacterial Lineages Involved in Benthic Carbon and Sulfur Cycling. Appl. Environ. Microbiol. 2015, 81, 6901–6914.
- Spring, S.; Bunk, B.; Sproer, C.; Rohde, M.; Klenk, H.P. Genome biology of a novel lineage of planctomycetes widespread in anoxic aquatic environments. Environ. Microbiol. 2018, 20, 2438–2455.
- Zhang, X.L.; Zhang, Q.Q.; Yang, A.J.; Hou, L.J.; Zheng, Y.L.; Zhai, W.D.; Gong, J. Incorporation of Microbial Functional Traits in Biogeochemistry Models Provides Better Estimations of Benthic Denitrification and Anammox Rates in Coastal Oceans. J. Geophys. Res. Biogeosci. 2018, 123, 3331–3352.
- Zhang, Y.; Ling, J.; Yang, Q.; Wen, C.; Yan, Q.; Sun, H.; Van Nostrand, J.D.; Shi, Z.; Zhou, J.; Dong, J. The functional gene composition and metabolic potential of coral-associated microbial communities. Sci. Rep. 2015, 5, 16191.
- Jiang, Y.F.; Ling, J.; Wang, Y.S.; Chen, B.; Zhang, Y.Y.; Dong, J.D. Cultivation-dependent analysis of the microbial diversity associated with the seagrass meadows in Xincun Bay, South China Sea. Ecotoxicology 2015, 24, 1540–1547.
- Park, S.; Yoshizawa, S.; Kogure, K.; Yokota, A. Rubricoccus marinus gen. nov., sp nov., of the family ‘Rhodothermaceae’, isolated from seawater. Int. J. Syst. Evol. Microbiol. 2011, 61, 2069–2072.
- Ugarelli, K.; Laas, P.; Stingl, U. The Microbial Communities of Leaves and Roots Associated with Turtle Grass (Thalassia testudinum) and Manatee Grass (Syringodium filliforme) are Distinct from Seawater and Sediment Communities, but Are Similar between Species and Sampling Sites. Microorganisms 2019, 7, 4.
- Deng, Y.; Jiang, Y.H.; Yang, Y.F.; He, Z.L.; Luo, F.; Zhou, J.Z. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113.
- Brodersen, K.E.; Nielsen, D.A.; Ralph, P.J.; Kuhl, M. Oxic microshield and local pH enhancement protects Zostera muelleri from sediment derived hydrogen sulphide. New Phytol. 2015, 205, 1264–1276.
- Martin, B.C.; Bougoure, J.; Ryan, M.H.; Bennett, W.W.; Colmer, T.D.; Joyce, N.K.; Olsen, Y.S.; Kendrick, G.A. Oxygen loss from seagrass roots coincides with colonisation of sulphide-oxidising cable bacteria and reduces sulphide stress. ISME J. 2018, 13, 707–719.
- Kuypers, M.M. Microbiology: A division of labour combined. Nature 2015, 528, 487–488.
- Radecker, N.; Pogoreutz, C.; Voolstra, C.R.; Wiedenmann, J.; Wild, C. Nitrogen cycling in corals: The key to understanding holobiont functioning? Trends Microbiol. 2015, 23, 490–497.
- Welsh, D.T. Nitrogen fixation in seagrass meadows: Regulation, plant-bacteria interactions and significance to primary productivity. Ecol. Lett. 2000, 3, 58–71.
- van Kessel, M.A.; Speth, D.R.; Albertsen, M.; Nielsen, P.H.; Op den Camp, H.J.; Kartal, B.; Jetten, M.S.; Lucker, S. Complete nitrification by a single microorganism. Nature 2015, 528, 555–559.
- Daims, H.; Lebedeva, E.V.; Pjevac, P.; Han, P.; Herbold, C.; Albertsen, M.; Jehmlich, N.; Palatinszky, M.; Vierheilig, J.; Bulaev, A.; et al. Complete nitrification by Nitrospira bacteria. Nature 2015, 528, 504–509.
- Schlesner, H. Blastopirellula. In Bergey’s Manual of Systematics of Archaea and Bacteria; Wiley: Hoboken, NJ, USA, 2015; pp. 1–13.
- More, K.D.; Giosan, L.; Grice, K.; Coolen, M.J.L. Holocene paleodepositional changes reflected in the sedimentary microbiome of the Black Sea. Geobiology 2019, 17, 436–448.
- Martin, B.C.; Alarcon, M.S.; Gleeson, D.; Middleton, J.A.; Fraser, M.W.; Ryan, M.H.; Holmer, M.; Kilminster, K. Root microbiomes as indicators of seagrass health. FEMS Microbiol. Ecol. 2019, 96, fiz201.

