 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Thomas Senoner | + 3231 word(s) | 3231 | 2021-08-25 05:49:33 | | | |
| 2 | Peter Tang | Meta information modification | 3231 | 2021-08-27 04:10:37 | | |
Video Upload Options
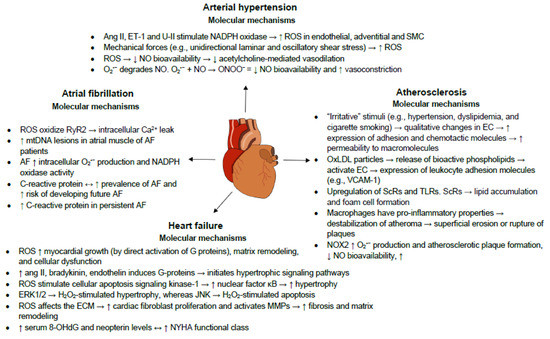
Cardiovascular diseases (CVD) are complex entities with heterogenous pathophysiologic mechanisms and increased oxidative stress has been viewed as one of the potential common etiologies. A fine balance between the presence of reactive oxygen species (ROS) and antioxidants is essential for the proper normal functioning of the cell. A basal concentration of ROS is indispensable for the manifestation of cellular functions, whereas excessive levels of ROS cause damage to cellular macromolecules such as DNA, lipids and proteins, eventually leading to necrosis and apoptotic cell death. CVD is the main cause of death worldwide with several conditions being affected by oxidative stress. Increased ROS lead to decreased nitric oxide availability and vasoconstriction, promoting arterial hypertension. ROS also negatively influence myocardial calcium handling, causing arrhythmia, and augment cardiac remodeling by inducing hypertrophic signaling and apoptosis. Finally, ROS have also been shown to promote atherosclerotic plaque formation.
1. Introduction
2. Endothelial Dysfunction in Cardiovascular Disease
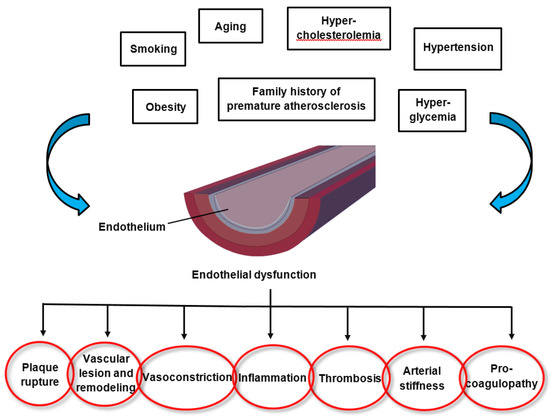
3. Oxidative Stress and Inflammation in Cardiovascular Diseases
4. Effects of Diet and Nutraceuticals on Oxidative Stress in Cardiovascular Diseases
|
Study Reference |
Diet/Nutraceutical(s) |
Studied Population |
Main Therapeutic Antioxidant Effects |
|---|---|---|---|
|
[47] |
Extra virgin olive oil |
Human and in vitro studies |
Stable platelet ROS generation, platelet and serum sNOX2-dp release, 8-iso-PGF2α-III formation, sVCAM1 and E-selectin levels and a NS ↓ in Vit E levels In vitro ↓ cell ROS and 8-iso-PGF2-III formation and NOX2 regulation |
|
[48] |
Dark chocolate (cocoa) |
Human study |
↓ platelet ROS and NOX2 activation, ↓ platelet production of 8-iso-PGF2a, ↑ platelet NOx levels after dark chocolate consumption in smokers |
|
Nuts |
In vitro, animal and human studies |
↓ lipid peroxidation, ↓ LDL oxidation, ↓ formation of TBARS, ↓ ROS-induced DNA strand scissions ↑ increase serum paraoxonase-1 and arylesterase activities ↓ DNA strand breaks in lymphocytes and 8-hydroxydeoxyguanosine urine concentrations |
|
|
Tea polyphenols |
In vitro, animal and human studies |
Scavenging of superoxide and other ROS, inhibition of lipid peroxidation ↑ plasma antioxidant capacity ↓ blood pressure, heart failure and atherosclerosis risk |
|
|
Flavonoids |
Animal and human studies |
↑ acetylcholine-induced nitric oxide production, ↓ acetylcholinesterase activity ↑ eNOS expression and GSH/GSSG ratio ↑ flow-mediated dilation, ↓ blood pressure ↓ NOx, iNOS expression and O2•− production ↓ lipid peroxidation and protein oxidation |
|
|
The Mediterranean diet |
Human studies |
↑ adherence to the MD associated with ↓ oxidative stress and inflammation and ↑ endothelial function and insulin sensitivity ↑ diet score associated with ↑ GSH/GSSG ratio |
|
|
PUFAs |
In vitro, animal and human studies |
↑ endothelial progenitor cells, ↓ endothelial microparticles inhibition of TLR and TNF-α pro-inflammatory signaling pathways, ↑ insulin sensitivity ↓ ROS-induced DNA damage, ↓ double-strand breaks, ↓ activation of kinases that initiate DNA damage response |
8-OHdG: 8-hydroxy-2′-deoxyguanosine; 8-iso-PGF2α-III: 8-iso-prostaglandin F2α; eNOS: endothelial nitric oxide synthase; GSH: glutathione; GSSG: glutathione disulfide; iNOS: inducible nitric oxide synthase; LDL: low-density lipoprotein; MD: Mediterranean diet; NOx: the nitric oxide metabolites nitrite and nitrate; NOX2: NADPH oxidase 2; NS: not significant; PUFAs: polyunsaturated fatty acids; ROS: reactive oxygen species; sVCAM1: vascular cell adhesion molecule 1; sNOX2-dp: soluble NOX isoform 2; TBARS: thiobarbituric acid reactive substances; TLR: toll-like receptor; TNF: tumor necrosis factor; Vit.: vitamin.
5. Benefits and Harms of Antioxidants
References
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Circ. Physiol. 2011, 301, H2181–H2190.
- Tahhan, A.S.; Sandesara, P.B.; Hayek, S.S.; Alkhoder, A.; Chivukula, K.; Hammadah, M.; Mohamed-Kelli, H.; O’Neal, W.T.; Topel, M.; Ghasemzadeh, N.; et al. Association between oxidative stress and atrial fibrillation. Heart Rhythm 2017, 14, 1849–1855.
- Baradaran, A.; Nasri, H.; Rafieian-Kopaei, M. Oxidative stress and hypertension: Possibility of hypertension therapy with antioxidants. J. Res. Med. Sci. 2014, 19, 358–367.
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42.
- Liochev, S.I. Reactive oxygen species and the free radical theory of aging. Free Radic. Biol. Med. 2013, 60, 1–4.
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421.
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15.
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495.
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748.
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics—2019 Update: A Report from the American Heart Association. Circulation 2019, 139, 2019.
- Huynh, D.T.N.; Heo, K.-S. Therapeutic targets for endothelial dysfunction in vascular diseases. Arch. Pharm. Res. 2019.
- Volobueva, A.; Grechko, A.; Yet, S.-F.; Sobenin, I.; Orekhov, A. Changes in Mitochondrial Genome Associated with Predisposition to Atherosclerosis and Related Disease. Biomolecules 2019, 9, 377.
- Al Disi, S.S.; Anwar, M.A.; Eid, A.H. Anti-hypertensive Herbs and their Mechanisms of Action: Part I. Front. Pharmacol. 2016, 6, 323.
- Godo, S.; Shimokawa, H. Endothelial Functions. Arter. Thromb. Vasc. Biol. 2017, 37, e108–e114.
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial Function and Dysfunction. Circulation 2007, 115, 1285–1295.
- Forstermann, U.; Munzel, T. Endothelial Nitric Oxide Synthase in Vascular Disease. Circulation 2006, 113, 1708–1714.
- Favero, G.; Paganelli, C.; Buffoli, B.; Rodella, L.F.; Rezzani, R. Endothelium and its alterations in cardiovascular diseases: Life style intervention. Biomed Res. Int. 2014, 2014, 801896.
- Hadi, H.A.R.; Carr, C.S.; al Suwaidi, J. Endothelial dysfunction: Cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005, 1, 183–198.
- Ghosh, A.; Gao, L.; Thakur, A.; Siu, P.M.; Lai, C.W.K. Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 2017, 24, 50.
- Durrant, J.R.; Seals, D.R.; Connell, M.L.; Russell, M.J.; Lawson, B.R.; Folian, B.J.; Donato, A.J.; Lesniewski, L.A. Voluntary wheel running restores endothelial function in conduit arteries of old mice: Direct evidence for reduced oxidative stress, increased superoxide dismutase activity and down-regulation of NADPH oxidase. J. Physiol. 2009, 587, 3271–3285.
- Pierce, G.L.; Lesniewski, L.A.; Lawson, B.R.; Beske, S.D.; Seals, D.R. Nuclear Factor-κB Activation Contributes to Vascular Endothelial Dysfunction via Oxidative Stress in Overweight/Obese Middle-Aged and Older Humans. Circulation 2009, 119, 1284–1292.
- Wang, M.; Zhang, J.; Jiang, L.Q.; Spinetti, G.; Pintus, G.; Monticone, R.; Kolodgie, F.D.; Virmani, R.; Lakatta, E.G. Proinflammatory Profile Within the Grossly Normal Aged Human Aortic Wall. Hypertension 2007, 50, 219–227.
- Lesniewski, L.A.; Durrant, J.R.; Connell, M.L.; Folian, B.J.; Donato, A.J.; Seals, D.R. Salicylate Treatment Improves Age-Associated Vascular Endothelial Dysfunction: Potential Role of Nuclear Factor B and Forkhead Box O Phosphorylation. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2011, 66, 409–418.
- Skoog, T.; Dichtl, W.; Boquist, S.; Skoglund-Andersson, C.; Karpe, F.; Tang, R.; Bond, M.G.; de Faire, U.; Nilsson, J.; Eriksson, P.; et al. Plasma tumour necrosis factor-α and early carotid atherosclerosis in healthy middle-aged men. Eur. Heart J. 2002, 23, 376–383.
- Dichtl, W.; Nilsson, L.; Goncalves, I.; Ares, M.P.; Banfi, C.; Calara, F.; Hamsten, A.; Eriksson, P.; Nilsson, J. Very low-density lipoprotein activates nuclear factor-kappaB in endothelial cells. Circ. Res. 1999, 84, 1085–1094.
- Dichtl, W.; Dulak, J.; Frick, M.; Alber, H.F.; Schwarzacher, S.P.; Ares, M.P.; Nilsson, J.; Pachinger, O.; Weidinger, F. HMG-CoA reductase inhibitors regulate inflammatory transcription factors in human endothelial and vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 58–63.
- Donato, A.J.; Pierce, G.L.; Lesniewski, L.A.; Seals, D.R. Role of NFkappaB in age-related vascular endothelial dysfunction in humans. Aging 2009, 1, 678–680.
- Anrather, J.; Racchumi, G.; Iadecola, C. NF-κB Regulates Phagocytic NADPH Oxidase by Inducing the Expression of gp91phox. J. Biol. Chem. 2006, 281, 5657–5667.
- Guzik, T.J.; Harrison, D.G. Endothelial NF-κB as a Mediator of Kidney Damage. Circ. Res. 2007, 101, 227–229.
- De Winther, M.P.J.; Kanters, E.; Kraal, G.; Hofker, M.H. Nuclear Factor κB Signaling in Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 904–914.
- Vasa, M.; Fichtlscherer, S.; Adler, K.; Aicher, A.; Martin, H.; Zeiher, A.M.; Dimmeler, S. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation 2001, 103, 2885–2890.
- Laufs, U.; Werner, N.; Link, A.; Endres, M.; Wassmann, S.; Jürgens, K.; Miche, E.; Böhm, M.; Nickenig, G. Physical Training Increases Endothelial Progenitor Cells, Inhibits Neointima Formation, and Enhances Angiogenesis. Circulation 2004, 109, 220–226.
- Hill, J.M.; Zalos, G.; Halcox, J.P.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating Endothelial Progenitor Cells, Vascular Function, and Cardiovascular Risk. N. Engl. J. Med. 2003, 348, 593–600.
- Barton, M. Prevention and endothelial therapy of coronary artery disease. Curr. Opin. Pharmacol. 2013, 13, 226–241.
- Watanabe, M.; Inukai, K.; Sumita, T.; Ikebukuro, K.; Ito, D.; Kurihara, S.; Ono, H.; Awata, T.; Katayama, S. Effects of telmisartan on insulin resistance in Japanese type 2 diabetic patients. Intern. Med. 2010, 49, 1843–1847.
- Khanicheh, E.; Mitterhuber, M.; Xu, L.; Haeuselmann, S.P.; Kuster, G.M.; Kaufmann, B.A. Noninvasive Ultrasound Molecular Imaging of the Effect of Statins on Endothelial Inflammatory Phenotype in Early Atherosclerosis. PLoS ONE 2013, 8, e58761.
- Liu, G.; Li, Y.; Gao, X.-G. MicroRNA-181a is upregulated in human atherosclerosis plaques and involves in the oxidative stress-induced endothelial cell dysfunction through direct targeting Bcl-2. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3092–3100.
- Xu, R.-H.; Liu, B.; Wu, J.-D.; Yan, Y.-Y.; Wang, J.-N. MiR-143 is involved in endothelial cell dysfunction through suppression of glycolysis and correlated with atherosclerotic plaques formation. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4063–4071.
- Li, P.; et al. Inhibition of Aberrant MicroRNA-133a Expression in Endothelial Cells by Statin Prevents Endothelial Dysfunction by Targeting GTP Cyclohydrolase 1 in Vivo. Circulation 2016, 134, 1752–1765.
- Loyer, X.; Potteaux, S.; Vion, A.C.; Guérin, C.L.; Boulkroun, S.; Rautou, P.E.; Ramkhelawon, B.; Esposito, B.; Dalloz, M.; Paul, J.L.; et al. Inhibition of MicroRNA-92a Prevents Endothelial Dysfunction and Atherosclerosis in Mice. Circ. Res. 2014, 114, 434–443.
- Pan, W.; Yu, H.; Huang, S.; Zhu, P. Resveratrol Protects against TNF-α-Induced Injury in Human Umbilical Endothelial Cells through Promoting Sirtuin-1-Induced Repression of NF-KB and p38 MAPK. PLoS ONE 2013, 11, e0147034.
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313.
- Cave, A.C.; Brewer, A.C.; Narayanapanicker, A.; Ray, R.; Grieve, D.J.; Walker, S.; Shah, A.M. NADPH Oxidases in Cardiovascular Health and Disease. Antioxid. Redox Signal. 2006, 8, 691–728.
- Brandes, R.P.; Weissmann, N.; Schröder, K. NADPH oxidases in cardiovascular disease. Free Radic. Biol. Med. 2010, 49, 687–706.
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435.
- Kalra, E.K. Nutraceutical-definition and introduction. AAPS PharmSci 2003, 5, 27–28.
- Carnevale, R.; Pignatelli, P.; Nocella, C.; Loffredo, L.; Pastori, D.; Vicario, T.; Petruccioli, A.; Bartimoccia, S.; Violi, F. Extra virgin olive oil blunt post-prandial oxidative stress via NOX2 down-regulation. Atherosclerosis 2014, 235, 649–658.
- Carnevale, R.; Loffredo, L.; Pignatelli, P.; Nocella, C.; Bartimoccia, S.; Di Santo, S.; Martino, F.; Catasca, E.; Perri, L.; Violi, F. Dark chocolate inhibits platelet isoprostanes via NOX2 down-regulation in smokers. J. Thromb. Haemost. 2012, 10, 125–132.
- Wijeratne, S.S.K.; Abou-Zaid, M.M.; Shahidi, F. Antioxidant Polyphenols in Almond and Its Coproducts. J. Agric. Food Chem. 2006, 54, 312–318.
- Chen, C.-Y.; Milbury, P.E.; Lapsley, K.; Blumberg, J.B. Flavonoids from Almond Skins Are Bioavailable and Act Synergistically with Vitamins C and E to Enhance Hamster and Human LDL Resistance to Oxidation. J. Nutr. 2005, 135, 1366–1373.
- Anderson, K.J.; Teuber, S.S.; Gobeille, A.; Cremin, P.; Waterhouse, A.L.; Steinberg, F.M. Walnut polyphenolics inhibit in vitro human plasma and LDL oxidation. J. Nutr. 2001, 131, 2837–2842.
- Shahidi, F.; Alasalvar, C.; Liyana-Pathirana, C.M. Antioxidant phytochemicals in hazelnut kernel (Corylus avellana L.) and hazelnut byproducts. J. Agric. Food Chem. 2007, 55, 1212–1220.
- Aksoy, N.; Aksoy, M.; Bagci, C.; Gergerlioglu, H.S.; Celik, H.; Herken, E.; Yaman, A.; Tarakcioglu, M.; Soydinc, S.; Sari, I.; et al. Pistachio intake increases high density lipoprotein levels and inhibits low-density lipoprotein oxidation in rats. Tohoku J. Exp. Med. 2007, 212, 43–48.
- Hatipoğlu, A.; Kanbağli, O.; Balkan, J.; Küçük, M.; Cevikbaş, U.; Aykaç-Toker, G.; Berkkan, H.; Uysal, M. Hazelnut oil administration reduces aortic cholesterol accumulation and lipid peroxides in the plasma, liver, and aorta of rabbits fed a high-cholesterol diet. Biosci. Biotechnol. Biochem. 2004, 68, 2050–2057.
- Kocyigit, A.; Koylu, A.A.; Keles, H. Effects of pistachio nuts consumption on plasma lipid profile and oxidative status in healthy volunteers. Nutr. Metab. Cardiovasc. Dis. 2006, 16, 202–209.
- Canales, A.; Benedí, J.; Nus, M.; Librelotto, J.; Sánchez-Montero, J.M.; Sánchez-Muniz, F.J. Effect of walnut-enriched restructured meat in the antioxidant status of overweight/obese senior subjects with at least one extra CHD-risk factor. J. Am. Coll. Nutr. 2007, 26, 225–232.
- Li, N.; Jia, X.; Chen, C.Y.O.; Blumberg, J.B.; Song, Y.; Zhang, W.; Zhang, X.; Ma, G.; Chen, J. Almond consumption reduces oxidative DNA damage and lipid peroxidation in male smokers. J. Nutr. 2007, 137, 2717–2722.
- Jia, X.; Li, N.; Zhang, W.; Zhang, X.; Lapsley, K.; Huang, G.; Blumberg, J.; Ma, G.; Chen, J. A pilot study on the effects of almond consumption on DNA damage and oxidative stress in smokers. Nutr. Cancer 2006, 54, 179–183.
- Graham, H.N. Green tea composition, consumption, and polyphenol chemistry. Prev. Med. 1992, 21, 334–350.
- Frei, B.; Higdon, J.V. Antioxidant Activity of Tea Polyphenols in Vivo: Evidence from Animal Studies. J. Nutr. 2003, 133, 3275S–3284S.
- Lambert, J.D.; Elias, R.J. The antioxidant and pro-oxidant activities of green tea polyphenols: A role in cancer prevention. Arch. Biochem. Biophys. 2010, 501, 65–72.
- Tang, G.-Y.; Zhao, C.N.; Xu, X.Y.; Gan, R.Y.; Cao, S.Y.; Liu, Q.; Shang, A.; Mao, Q.Q.; Li, H.B. Phytochemical Composition and Antioxidant Capacity of 30 Chinese Teas. Antioxidants 2019, 8, 180.
- Oyama, J.-I.; Shiraki, A.; Nishikido, T.; Maeda, T.; Komoda, H.; Shimizu, T.; Makino, N.; Node, K. EGCG, a green tea catechin, attenuates the progression of heart failure induced by the heart/muscle-specific deletion of MnSOD in mice. J. Cardiol. 2017, 69, 417–427.
- Liu, L.; Zhao, W.; Liu, J.; Gan, Y.; Liu, L.; Tian, J. Epigallocatechin-3 gallate prevents pressure overload-induced heart failure by upregulating SERCA2a via histone acetylation modification in mice. PLoS ONE 2018, 13, e0205123.
- Chen, X.Q.; Hu, T.; Han, Y.; Huang, W.; Yuan, H.B.; Zhang, Y.T.; Du, Y.; Jiang, Y.W. Preventive effects of catechins on cardiovascular disease. Molecules 2016, 21, 1759.
- Peng, X.; Zhou, R.; Wang, B.; Yu, X.; Yang, X.; Liu, K.; Mi, M. Effect of green tea consumption on blood pressure: A meta-analysis of 13 randomized controlled trials. Sci. Rep. 2014, 4, 6251.
- Kuriyama, S.; Shimazu, T.; Ohmori, K.; Kikuchi, N.; Nakaya, N.; Nishino, Y.; Tsubono, Y.; Tsuji, I. Green Tea Consumption and Mortality Due to Cardiovascular Disease, Cancer, and All Causes in Japan. JAMA 2006, 296, 1255.
- Clark, J.L.; Zahradka, P.; Taylor, C.G. Efficacy of flavonoids in the management of high blood pressure. Nutr. Rev. 2015, 73, 799–822.
- Kay, C.D.; Hooper, L.; Kroon, P.A.; Rimm, E.B.; Cassidy, A. Relative impact of flavonoid composition, dose and structure on vascular function: A systematic review of randomised controlled trials of flavonoid-rich food products. Mol. Nutr. Food Res. 2012, 56, 1605–1616.
- Fairlie-Jones, L.; Davison, K.; Fromentin, E.; Hill, A.M. The Effect of Anthocyanin-Rich Foods or Extracts on Vascular Function in Adults: A Systematic Review and Meta-Analysis of Randomised Controlled Trials. Nutrients 2017, 9, 908.
- McCullough, M.L.; Peterson, J.J.; Patel, R.; Jacques, P.F.; Shah, R.; Dwyer, J.T. Flavonoid intake and cardiovascular disease mortality in a prospective cohort of US adults. Am. J. Clin. Nutr. 2012, 95, 454.
- Schwingshackl, L.; Morze, J.; Hoffmann, G. Mediterranean diet and health status: Active ingredients and pharmacological mechanisms. Br. J. Pharmacol. 2019.
- Schwingshackl, L.; Hoffmann, G. Mediterranean dietary pattern, inflammation and endothelial function: A systematic review and meta-analysis of intervention trials. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 929–939.
- Dai, J.; Jones, D.P.; Goldberg, J.; Ziegler, T.R.; Bostick, R.M.; Wilson, P.W.; Manatunga, A.K.; Shallenberger, L.; Jones, L.; Vaccarino, V. Association between adherence to the Mediterranean diet and oxidative stress. Am. J. Clin. Nutr. 2008, 88, 1364–1370.
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T. Jr.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N. Engl. J. Med. 2019, 380, 11–22.
- Wu, S.-Y.; Mayneris-Perxachs, J.; Lovegrove, J.A.; Todd, S.; Yaqoob, P. Fish-oil supplementation alters numbers of circulating endothelial progenitor cells and microparticles independently of eNOS genotype. Am. J. Clin. Nutr. 2014, 100, 1232–1243.
- Mozaffarian, D.; Wu, J.H.Y. Omega-3 fatty acids and cardiovascular disease: Effects on risk factors, molecular pathways, and clinical events. J. Am. Coll. Cardiol. 2011, 58, 2047–2067.
- Serhan, C.N.; Chiang, N.; van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361.
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698.
- Sakai, C.; Ishida, M.; Ohba, H.; Yamashita, H.; Uchida, H.; Yoshizumi, M.; Ishida, T. Fish oil omega-3 polyunsaturated fatty acids attenuate oxidative stress-induced DNA damage in vascular endothelial cells. PLoS ONE 2017, 12, e0187934.
- Biesalski, H.K.; Grune, T.; Tinz, J.; Zöllner, I.; Blumberg, J.B. Reexamination of a meta-analysis of the effect of antioxidant supplementation on mortality and health in randomized trials. Nutrients 2010, 2, 929–949.
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011, 51, 327–336.
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84.
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762.
- Halliwell, B. Free radicals and antioxidants: Updating a personal view. Nutr. Rev. 2012, 70, 257–265.

