1. Introduction
A variety of cardiovascular diseases have been shown to be associated, at least partially, with an excess production of reactive oxygen species (ROS) [
1,
2,
3,
4]. ROS constitute both oxygen free radicals, such as superoxide, hydroxyl radicals, and peroxyl radicals, as well as non-radicals, such as hydrogen peroxide, hypochlorous acid, and ozone [
5]. In most cell types, mitochondria are the main drivers of intracellular oxidant production, while other relevant sources are nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (summarized as the NOX family of enzymes). Apart from that, numerous other enzymes such as xanthine oxidase, nitric oxide synthase, cyclooxygenases, cytochrome P450 enzymes, and lipoxygenases as well as other cell organelles, like the peroxisome and endoplasmic reticulum, contribute to intracellular ROS production [
6]. Proteins, lipids and DNA are the primary cellular structures affected by ROS and reactive nitrogen species (RNS). The generation of molecular oxygen in the form of ROS is a natural part of aerobic life. Indeed, basal levels of ROS are essential for the manifestation of various cellular functions, such as signal transduction pathways, defense against invading microorganisms, gene expression and the promotion of growth or death [
7]. In spite of the crucial relevance of redox reactions, dysregulation of oxidant signaling may cause or accelerate a host of pathological conditions, such as the rate of aging. However, the body is equipped with protective measures against ROS via enzymatic (e.g., superoxide dismutase (SOD), catalase (CAT), peroxiredoxin (Prx) and glutathione peroxidase (GSH-Px)) as well as non-enzymatic compounds (e.g., tocopherol/vitamin E, beta-carotene, ascorbate, glutathione (GSH), and nicotinamide (NAM)) [
8]. Newer research tools allow the investigation of redox signaling pathways in adequate chemical detail, and it has become clear that redox processes are as important in biology as phosphorylation-dephosphorylation reactions, or central mechanisms responsible for controlling the genome and epigenome, such as acetylation–deacetylation and methylation–demethylation [
9]. However, the analysis of redox systems is challenging due to the substantial subcellular differences in redox potential and the short lifespan of ROS. The discovery of numerous biomarkers of oxidative stress has facilitated the measurement of ROS; however, their clinical use still needs to be validated given the vast diversity in oxidative stress between different diseases. Genomics, epigenomics, and exposomics along with methodologies for redox imaging, redox proteomics, and redox metabolomics, will improve our understanding of health and disease processes within entire biological systems. Furthermore, the emerging big data and artificial intelligence era will provide us with new opportunities for the development of oxidative stress knowledge bases and paves the way for a more personalized redox medicine [
9].
In 2016, ≈17.6 million deaths were attributed to cardiovascular diseases (CVD) globally, which amounted to an increase of 14.5% from 2006 [
10]. CVD is currently the leading cause of death, and it claims more lives each year than cancer and chronic lung disease combined. Coronary heart disease (CHD) represents the most common CVD [
10]. In the coming decades, with an aging population and increased incidence of obesity and diabetes, the burden and medical costs of CVD are anticipated to significantly increase worldwide. Although significant efforts have been made to enlighten pathophysiologic mechanisms governing the initiation and progression of CVD, still much work has to be done [
11,
12,
13]. As such, a better understanding of the biomolecular mechanisms and their clinical consequences is urgently needed to reduce the burden of CVD, and this poses a serious challenge in medicine.
2. Endothelial Dysfunction in Cardiovascular Disease
The endothelium is a highly active monolayer that plays important roles in modulating vascular tone, cellular adhesion, thromboresistance, smooth muscle cell proliferation, and vessel wall inflammation. This is achieved by the production and release of several endothelium-derived relaxing factors, including vasodilator prostaglandins, nitric oxide (NO), and endothelium-dependent hyperpolarization factors, as well as endothelium-derived contracting factors. These vasoactive molecules that relax or constrict the vessel play a direct role in the balance of tissue oxygen supply, long-term organ perfusion, remodeling of vascular structures, and metabolic demand by regulation of vessel tone and diameter [
14,
15]. Endothelial cells dispose of an enzyme to fight vascular disease, namely endothelial nitric oxide synthase (eNOS), which generates the vasoprotective molecule NO. This molecule diffuses to the vascular smooth muscle cells, activates soluble guanylyl cyclase and increases cyclic guanosine monophosphate (cGMP) [
16]. NO can also inhibit leukocyte adhesion to the vessel wall which represents an early event in the development of atherosclerosis; therefore, NO may protect against the onset of atherogenesis. Furthermore, NO is also involved in the inhibition of platelet aggregation and adhesion, both of which protect smooth muscle cells from exposure to platelet-derived growth factors. These mechanisms can lead to fibrous plaque formation; therefore, NO also prevents a later step in atherogenesis. NO suppresses key processes in vascular lesion formation and thus probably represents the most important antiatherogenic defense principle in the vasculature [
16].
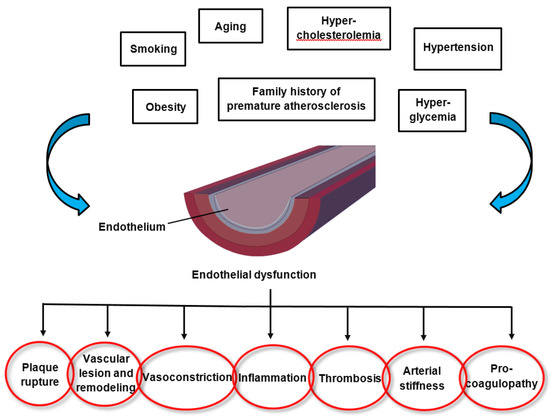
The pathomechanisms of endothelial dysfunction involve a diminished production and/or availability of NO, and a disproportion between the endothelium-derived vasodilators and vasoconstrictors. Several traditional cardiovascular risk factors are associated with alteration in endothelial function such as smoking, sedentary and incorrect lifestyle, aging, hypercholesterolemia, arterial hypertension, hyperglycemia, and a family history of premature atherosclerotic disease (
Figure 1) [
17,
18]. This leads to chronic inflammation, resulting in an increase in vasoconstrictor and prothrombotic products and diminished antithrombotic factors, in addition to abnormal vasoreactivity, all of which elevate the risk of cardiovascular events. Indeed, endothelial dysfunction has also been linked with obesity, elevated C-reactive protein, and chronic systemic infection [
18]. Oxidative stress and inflammation are the main drivers of endothelial dysfunction. Several oxidative enzyme systems such as NADPH oxidase, xanthine oxidase, cyclooxygenases, lipoxygenases, myeloperoxidases, cytochrome P450 monooxygenase, uncoupled NOS, and peroxidases lead to the inactivation of NO, which represents a critical mechanism leading to endothelial dysfunction through an elevated level of superoxide anion (O
2•−) [
19]. Both NADPH oxidase and eNOS uncoupling (i.e., the generation of ROS through eNOS as part of endothelial activation) act as important sources of O
2•− that give rise to vascular oxidative stress. Inhibition of NADPH oxidase has been established as a key molecular mechanism leading to reduced arterial oxidative stress and normalization of endothelial dysfunction in mice [
20]. Inflammation has been shown in many studies to play a role in endothelial dysfunction that underlies the pathogenesis of CVD, obesity and type 2 diabetes mellitus. Both in rodents and humans, elevated levels of pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1beta (IL-1 β), interleukin-6 (IL-6), and interferon gamma (IFN-γ) have been observed in age-related endothelial dysfunction, mainly via the activation of the nuclear factor-kappa B (NF-κB) pathway [
21,
22,
23,
24,
25,
26]. NF-κB is an important transcription factor that regulates the gene expression of factors responsible for cell adhesion, proliferation, inflammation, redox status, and tissue specific enzymes. It is expressed in all cell types and plays a major role in the promotion of CVD through the transcription of pro-inflammatory, pro-adhesion and pro-oxidant genes [
27]. The NF-κB pathway can be activated by a variety of stimuli including inflammatory cytokines, ROS, lipids and mechanical forces acting on the vascular endothelial wall. Upon activation, transmembrane receptors are stimulated which trigger intracellular signaling pathways, culminating in the activation of a kinase (IκK) mediated phosphorylation and degradation of the inhibitor of NF-κB (IκB). Subsequently, the NF-κB heterodimer (p65/p50 subunits and, perhaps, p65, RelB, c-Rel, p50 and p52) translocates to the nucleus, where it binds to promoters of gene targets. Several other pro-inflammatory molecules have been associated with endothelial dysfunction, such as IL-6, TNF-α, monocyte chemoattractant protein 1 (MCP-1), receptor for advance glycation endproducts (RAGE) and the pro-oxidant enzyme NADPH oxidase, and all predispose the vasculature to a “proatherogenic” phenotype [
28,
29,
30].
Figure 1. Factors altering endothelial function and the consequences of endothelial dysfunction.
The endothelium has an endogenous capacity for repair, which occurs via two mechanisms. Lost and damaged cells can be replaced by locally replicating mature endothelial cells. However, this repair mechanism is insufficient in the presence of risk factors and loss of endothelial integrity would rapidly ensue. Circulating endothelial progenitor cells represent an alternative mechanism for maintenance and repair of the endothelium; they are recruited from the bone marrow. These cells circulate in the peripheral blood and have the ability to differentiate into mature cells with endothelial characteristics [
15]. Indeed, factors that have been shown to have a positive impact on endothelial function, such as exercise and statins, have also been shown to have a potent positive effect on the mobilization of endothelial progenitor cells [
31,
32]. The importance of the balance between exposure to risk factors and the efficiency of endothelial repair has been underscored by the observation that subjects with increased numbers of circulating endothelial progenitor cells have preserved endothelial function, despite exposure to high levels of risk factors [
33].
Endothelial therapy can be viewed as a two-step approach. The best treatment for diseases is preventing the disease from occurring in the first place. Therefore, the first approach is disease prevention through increased awareness and control of cardiovascular risk factors by nonpharmacological measures such as lifestyle optimization (e.g., a healthy diet, physical exercise, maintaining a normal body weight). The second approach is targeted (pharmacological) therapy directed at preserving or restoring the function of already impaired endothelial cells in order to defer disease progression, promote disease stabilization, improve overall quality of life, reduce disability and health costs, and ultimately increase survival [
34]. The aim of pharmacological treatment is reestablishing endothelial cell homeostasis (e.g., statin therapy to reduce low-density lipoprotein (LDL) cholesterol levels; antidiabetics to reduce blood glucose levels; antihypertensives to normalize blood pressure; heart failure therapy to amend myocardial and vasomotor function). Certain drugs may have some additional, endothelium-protective off-target effects, such as statins by reducing inflammation and some angiotensin antagonists also having metabolic (antidiabetic) effects [
35,
36]. Researchers are currently exploring new pathogenetic targets to improve vascular dysfunction, including anti-inflammatory agents, therapies based on microRNAs and epigenetic mechanisms. MicroRNAs (miRNAs) have been demonstrated to play a pivotal role during atherosclerotic plaque formation. Indeed, it has been established that both miR-143 and miR-181a are upregulated in human atherosclerotic plaques [
37,
38]. Hydrogen peroxide (H
2O
2) treatment induces an increase in miR-181a levels, while inhibition of miR-181a leads to increased resistance to H
2O
2, thus implying that miR-181a is involved in the oxidative stress-induced endothelial cell dysfunction [
37]. MiR-133a may represent an additional target for preventing cardiovascular disease. Studies have demonstrated that the inhibition of aberrant miR-133a by lovastatin prevents endothelial dysfunction by targeting GTP cyclohydrolase 1, a critical enzyme for eNOS uncoupling in endothelial dysfunction [
39]. Finally, inhibition of miR-92a, an important regulator of endothelial proliferation and angiogenesis after ischemia, leads to reduced endothelial inflammation, decreased plaque size, and a more stable lesion phenotype [
40]. In recent years, emerging evidence has arisen that epigenetic pathways might also play an important role in endothelial dysfunction. Resveratrol, a member of the polyphenol group, is produced by several plants in response to injury and protects against pathological processes through the suppression of elevated levels of proinflammatory cytokines in macrophages. Increased TNF-α-induced CD40 expression has been shown to modify the expression levels of specific adhesion molecules, thus boosting the inflammatory response. Resveratrol treatment was able to attenuate the enhanced CD40 expression triggered by TNF-α stimulation. Furthermore, resveratrol suppressed TNF-α-triggered ROS via potentiating the activity of sirtuin 1 (a histone deacetylase involved in suppressing inflammation), thus protecting cells from damage generated by inflammatory factors [
41].