 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Simona Felletti | + 3349 word(s) | 3349 | 2021-08-15 08:55:54 | | | |
| 2 | Conner Chen | Meta information modification | 3349 | 2021-09-01 10:38:29 | | |
Video Upload Options
Peptides are organic polymers composed of 2–50 amino acids linked to each other by means of covalent amide (=peptide) bonds. The composition, length and sequence of the amino acid chain have a dramatic influence on the activity of the peptide itself, for example in the human body. Peptides are called bioactive if they have a beneficial impact on body functions, on biological processes and, as a consequence, on health. The main production methods to obtain peptides are enzymatic hydrolysis, microbial fermentation, recombinant approach and, especially, chemical synthesis.
1. Methods of Production (Upstream Processing)
1.1. Enzimatic Hydrolysis and Microbial Fermentation
Food is a valuable source of amino acids and peptides. For example, proteins contained in food can release peptides with bioactive functions during their fermentation or when exposed to enzymes with hydrolytic activity. The outcome of the hydrolysis, namely the type of peptides produced starting from a single parent protein, depends on many experimental factors (type of enzyme or microorganism used, combination of vitro enzymatic hydrolysis with microbial fermentation, etc.). Therefore, the number of bioactive peptides obtainable from food proteins is essentially unlimited [2]. Dairy products, for instance, are an important source of proteins in the human diet and, from them, a number of bioactive peptides can be obtained. Peptides with antihypertensive, antibacterial and immunomodulatory activity have been released by casein and by whey proteins using pepsin, trypsin and chymotripsin as enzymes [3][4][5][6]. Additionally, the enzyme thermolysin has been employed to obtain hypotensive peptides from other kind of foods, such as corn and porcine skeletal muscle [7][8][9][10]. The traditional mode to perform hydrolysis of proteins is to operate in batch, which means through a discontinuous process inside a reactor. This method, anyway, has resulted to be less efficient than continuous methods employing enzymatic membrane reactors, where protein hydrolysis, product collection and catalyst recovery happen in the same unit [11][12].
Dairy products can release bioactive peptides also when subjected to the action of particular bacteria that trigger the fermentation of this kind of foods. For example, it has been demonstrated that Lactobacillus helveticus, Enterococcus faecalis, yoghurt and cheese bacteria and Lactic Acid bacteria can hydrolyze milk proteins to produce peptides with ACE-inhibitory activity (antihypertensive) [5][13][14][15][16][17][18]. Similarly, peptide with this kind of bioactivity were produced from chicken meat, using Aspergillus protease [19].
Another kind of bioactivity that was observed in protein hydrolysates deriving from beef meat is the antioxidant one, especially against lipid oxidation. The employment of bioactive peptides as antioxidant additives in food could be pivotal in the substitution of artificial antioxidants, whose potential health risks are already recognized [20][21].
1.2. Chemical Synthesis
Despite the success of other production methods, the technique of choice to produce small to medium peptides, especially for pharmaceutical applications, is still chemical synthesis [22]. The main reasons are two: firstly, a synthesis method is developed starting from standard and well-established procedures, so its development is less complex and less time-consuming. Secondly, in the synthetic approach, differently than recombinant approach, modified amino acids can also be incorporated in the peptide chain. These characteristics make chemical synthesis the preferred technique for peptide production.
Chemical synthesis can be performed either in liquid- or in solid-phase. Both strategies are based on similar reaction mechanisms, where amino acids and/or fragments of the desired peptide are added successively to the mixture, to react with the growing chain. In Solid Phase Peptide Synthesis (SPPS), firstly developed by Merrifield [23], one N-protected amino acid reacts with the peptide chain, which is anchored to a solid support (a resin) and, after that, the terminal amino acid is deprotected. Then, the following amino acid undergoes the same procedure. Functionalities on the aminoacid side chain need to be protected as well, in order to avoid side reactions. After the procedure is terminated and all the amino acids have reacted, the peptide is released from the resin during the cleavage step. A scheme representing the Solid Phase Peptide Synthesis is depicted in Figure 1.
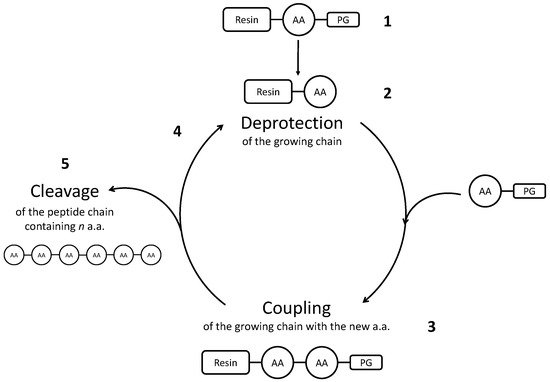
Figure 1. Scheme representing Solid Phase Peptide Synthesis. (1) An amino acid (AA) is protected on the functional group that must not react, and it is bonded to an insoluble resin. Then, its protecting group (=PG) is removed (2), so that the following amino acid, which in turn is protected, can bond to the growing chain (3). Successively, also the second amino acid is deprotected, to add the third one (4). After all the amino acids have been added, the peptide chain is recovered from the synthesis mixture with the step called cleavage (5).
The presence of the solid support allows one to recover the product simply by filtration: in this lies the reason of the success of synthesis in solid phase. Moreover, the process can be automated [24][25][26][27][28]. Currently, huge efforts are being made in order to make the synthetic processes as green as possible, by introducing the use of protecting groups and alternative solvents more sustainable than the traditional ones [29][30][31].
Liquid phase synthesis plays an important role in the manufacturing of short peptides (up to 10 amino acids). Recently, this approach gained importance for the manufacturing of longer peptides through the coupling of its previously synthesized fragments [32].
1.3. Recombinant Approach
This technique is the preferred one to produce peptides containing only natural amino acids on large scale. Compared to isolation from proteins and chemical synthesis, recombinant approach represents the most cost-effective and green way for large-scale peptide manufacturing. Particularly, Escherichia coli is the most widely used host. With this genetic engineering process, nevertheless, only peptides containing natural amino acids can be produced. Moreover, the biotechnological process requires great research efforts to develop a suitable procedure, and it is also time-consuming. Generally, the steps followed are: selection of an appropriate expression system, construction of expression vectors, development of the bioprocess. The operating conditions can be tailored for the specific product considered [24][26][33].
2. Purification Techniques (Downstream Processing)
None of the aforementioned upstream methods to obtain the peptide of interest leads to a single product. Actually, a series of impurities are produced together with the target. They must be removed during the downstream step of manufacturing. The reason is that every impurity could potentially exert adverse biological activity on the human body. Therefore, very strict purity requirements are applied to pharmaceuticals.
Different purification methods have been developed, with their own advantages and disadvantages. The purification strategy must be evaluated for every single case, in light of imposed requirements that, in turn, vary depending on the particular application [34].
If a peptide is produced through hydrolysis, for instance, it can be separated by means of ultrafiltration from the enzyme employed during the process and from other protein residues with higher molecular mass. Generally, in this case, the membranes of choice have a low molecular mass cut-off and the size of their pores depends on the molecular weight of the desired peptide. Anyway, the main disadvantage of this technique is the poor selectivity of the membrane [35][36].
A technique particularly useful in case of separation of charged peptides and proteins is IsoElectroFocusing (IEF), which is based on the same separation principles as electrophoresis. The sample is injected in a chamber where an electric field is applied, in presence of a pH gradient. Acidic species move towards the anode and basic ones towards the cathode. When a species reaches a zone with pH identical to its isoelectric point, it stops migrating. Then it can be moved to a detection windows to be identified. Therefore, IEF separates analytes depending on their isoelectric point [37]. Several modes of IEF have been developed, some of which can be used on analytical while others on preparative scale, which is the case of IEF in solution [38] or in a cellulose-based separation medium [39]. Anyway, IEF lies outside the main topic of this review and therefore it will not be treated any further.
A common purification issue is the separation of a complex mixture of peptides deriving from solid-phase synthesis. The separation can only be done after cleavage and it is frequently challenging because impurities may differ from the target peptide by a single amino acid or a single functional group, resulting in very similar chemical properties [40]. Typical by-products due to solid phase synthesis include peptides with one amino acid that did not react or reacted in the wrong position, peptides with side-chain modification (oxidation, deamidation, epimerization, alkylation, ring closure or opening, incomplete deprotection) and truncated peptides [41][42].
Chromatography is the most suitable technique for the purification of valuable products, which is the case of pharmaceuticals, where high resolution and selectivity are required [43][44]. This technique allows one to obtain very high efficiency in the separation of complex mixtures, where the components have similar chemical properties. It is flexible and adaptable since a wide selection of stationary and mobile phases to choose from is available. Additionally, a number of well established chromatographic methods have already been developed at industrial level and are available for practical biopharmaceutical applications [34]. A disadvantage of chromatographic methods is that it is difficult to handle viscous mixtures, which cause increase in the backpressure; moreover, organic solvents are used almost always as mobile phase, and this poses environmental concerns regarding their toxicity and disposal [45]. Anyway, chromatography remains the technique of choice for the purification of biomolecules at laboratory, preparative and industrial level. Currently, industrial processes for biopharmaceuticals employ almost exclusively chromatography both for capture and for polishing steps.
Due to the complexity of the peptide mixtures, generally a combination of chromatographic techniques based on different separation principles is required to improve the resolution power [46]. Therefore, at least two different chromatographic modes are applied consecutively, either online or offline, resulting in a multidimensional separation (e.g., two-dimensional liquid chromatography, 2D-LC). A separation performed on the basis of different types of interactions is often referred to with the term “orthogonality” [47], meaning that the two dimensions of the separation can remove different impurities. For example, ion-exchange, HILIC and reversed-phase chromatography separate the analytes depending on different features (charge, hydrophilicity and hydrophobicity) and therefore they can be used as dimensions orthogonal to each other [48]. Otherwise, also reversed-phase in acidic conditions and in basic conditions can be considered to be orthogonal separation methods and have shown very good results in terms of peak capacity when applied to purifications of peptide mixtures [49]. In the case where the two chromatographic modes are coupled offline, the product eluting from the first column is collected and then re-analyzed in the second dimension. This approach is quite labor-intensive and time-consuming. On the other side, in online multidimensional chromatography the product eluting from the first column is immediately injected into the second column, and this allows one to speed up a lot the analysis time, but requires compatibility between the solvents used in the two dimensions. For preparative scale purification, the heart-cutting mode is generally the preferred choice, meaning that only the peak of interest is further separated in the second dimension [50]. On the contrary, for analytical scale analysis, meaning to identify components in the mixture, comprehensive multidimensional separations are usually performed, where the eluent from the first dimension is injected into the second column over the entire first separation time [51]. Generally, mass spectrometry is coupled to multidimensional chromatography, especially at the outlet of the second column [52].
Multidimensional chromatography is based on the same principles as one-dimensional chromatography and therefore will not be further discussed. In the next paragraphs, different modes of chromatography will be considered in detail. Additionally, different purification techniques described in this Section and in the next ones are summarized in Table 1.
Table 1. In this table, the main techniques employed for peptide purification and their interaction mechanisms are summarized.
| Purification or | Mechanism |
|---|---|
| Identification Method | |
| Ultrafiltration | Target peptide is separated from other species depending on their size |
| IsoElectroFocusing (IEF) | Peptides are separated on the basis of their isoelectric point through an electric field and a pH gradient |
| Single-column chromatography | Different modes of chromatography have been developed, depending on the chemical features of the analytes:
|
| Multicolumn chromatography | Combination of two or more orthogonal chromatographic modes applied consecutively |
| MCSGP | Same separation principles as single-column chromatography but with the use of two or more identical columns. The performance parameters increase due to internal recycling of impure fractions into the system. |
2.1. Reversed-Phase Liquid Chromatography (RP-LC)
The mode of chromatography most frequently encountered when it comes to the purification of peptide and protein mixtures is reversed-phase liquid chromatography [53]. This technique separates the analytes depending on their hydrophobic properties [35]. Generally, C18 ligands are the most employed stationary phases in RP-LC, but occasionally C8 or even C4 ligands have shown better retentive characteristics, especially in case of very hydrophobic peptides. In other cases, also monolithic, poly(styrene-divinylbenzene)-based columns have been used [54]. All these stationary phases are able to distinguish diastereomers (peptide epimers for instance) but not enantiomers.
In RP-LC, the retention of macromolecules, such as peptides, decreases drastically with the content of organic modifier [27][55][56]. Therefore, it is recommended for macromolecules to use gradient elution, which also contributes to improve the separation of the target peptide from its product-related impurities, since species with similar structure can show very different adsorption behaviour at a given mobile phase composition [28]. Several peptide mixtures have been separated by means of gradient elution in RP-LC, such as insulin from its main degradation product (A21-desamido insulin) [57] and octreotide from impurities [28]. Using a very shallow gradient (0.1% ACN per min), Harris and coworkers [58] managed to purify mixtures of different synthetic polypeptides, with length ranging from 23 to 51 amino acids, containing closely eluting impurities. Sample amounts varying from 145 to 900 mg of peptide mixture could be purified with this one-step chromatographic method, including peptides modified with non-proteinogenic substituents (e.g., biotin, carboxyfluorescein); the purities reached were almost always above 95%.
Besides ACN and other more eco-friendly solvents have been tested and compared to it. Ethanol, for instance, has shown elution strength and separation characteristics similar to ACN during the separation of three peptides (bradykinin, angiotensin II, angiotensin I), thus resulting to be a promising candidate to substitute ACN in some cases [59].
Acidic ion-pairing agents (trifluoroacetic acid or formic acid) are often added to the mobile phase to pair with basic amino acids, that are positively charged at acidic pH, improving peak shape [60][61]. A study demonstrated that TFA concentration affects the recovery of the peptide or protein: at low TFA concentrations, below 0.05%, strong ion-exchange interactions can establish between analytes and hydrolyzed silanols, thus causing peak broadening [62].
Mazzoccanti et al. [63] developed a d-ERRP (dynamic Electrostatic-Repulsion Reversed-Phase) method (a variation of the classical static ERRP) based on the repulsion between the basic peptide and a hydrophobic ion-pairing agent adsorbed on the alkyl chains of the stationary phase, both positively charged at acidic pH. The hydrophobic agent used in that research was tetrabutylammonium, dissolved in the mobile phase together with TFA. This innovative chromatographic mode was successfully applied to the purification of glucagon (containing 29 amino acids) from its epimer [D-His]1-Glu and other four critical synthetic impurities deriving from deamidation or racemization of some amino acids. This technique is called “dynamic” because the repulsion is generated by the flow of the mobile phase, in opposition to static ERRP that will be discussed later.
A remarkable advantage of RP-LC over other chromatographic modes is that, thanks to the solvents employed, it can be easily coupled with Mass Spectrometry, a detection technique very popular for the characterization of macromolecules such as peptides and proteins [64]. This technique has allowed one, for instance, to separate bioactive peptides and tryptic digests of different proteins in RP-LC under both acidic and alkaline conditions, using trifluoroacetic acid or a buffer made of triethylamine and acetic acid respectively, and applying a gradient of ACN [54]. The detectability of peptides in conditions of full-scan negative-ion electrospray ionization mass spectrometry after the separation at high pH was two to three times lower with respect to their detectability in conditions of full-scan positive-ion electrospray ionization mass spectrometry after the separation at low pH.
2.2. Ion-Exchange Chromatography (IEX)
In ion-exchange chromatography, the separation mechanism is based on the electrostatic interactions between the opposite charges of analytes and stationary phase. Separation is modulated by the amount of competitive ions present in the mobile phase [35]. This technique is particularly suitable when dealing with peptide purification, because most peptides have a net charge that can be varied depending on the pH of the mobile phase. At acidic pH, carboxylate groups and basic residues (Arg, Hys, Lys) are mainly protonated. Therefore, IEX is often used to characterize charge variants of peptides and proteins, even though lately it has been more and more replaced by RP-LC because of its incompatibility with MS detection [65].
To identify peptide modifications such as deamidation or acetylation, that are detected with difficulty through RP-LC, IEX reveals to be a good choice. Moreover, this technique is able to distinguish between analytes with similar hydrophobicity. For example, using a strong cation-exchange column (Luna SCX containing a phenyl sulfonic acid exchanger) with salt gradient (namely, potassium chloride) it was possible to separate bradykinin variants differing only slightly in hydrophobicity or only by one charge [66].
Crimmins [67] verified that, during the IEX separation of a complex mixture of synthetic peptides positively charged from +1 to +7 at pH 3, the order of elution followed the order of charge monotonically; the most retained compound was the peptide with charge +7 (see Figure 2). In that case, the stationary phase used was a sulfoethyl aspartamide (a hydrophilic strong-cation exchange adsorbent), whereas mobile phases were MPA = 5 mM sodium phosphate pH 3, 25% acetonitrile (ACN), and MPB = 5 mM sodium phosphate, 500 mM NaCl pH 3, 25% acetonitrile. At low levels of ACN, retention and selectivity are mainly governed by the presence of basic amino acids and positive charges contained in the peptide. The same stationary phase has been tested by different research groups to analyze several peptides (up to 50 in a single study, ranging from 5 to 20 amino acid residues) [68][69][70]. However, the elution order does not always follow the order of charge. By analysing with the same method described above the peptide fragments produced by digestion of myoglobin, it was noted an inversion of the elution order with respect to the charge order [71]. This behaviour was attributed to the overall hydrophobicity of the peptide and to the fact that, for steric reasons, the charged residues do not interact simultaneously with the ion-exchange resin. Probably, there is a limit related to peptide molecular weight above which the monotonic relation between the elution order and the global charge of the analyte is no longer valid.
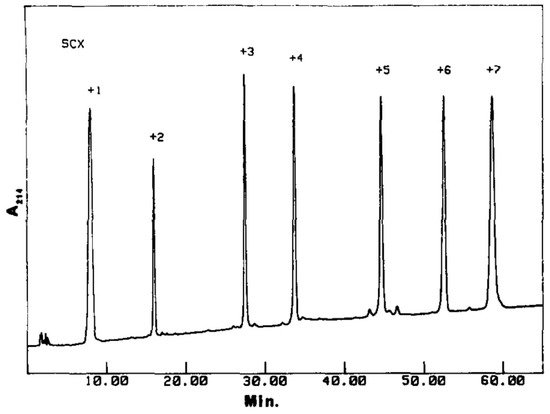
Figure 2. Separation of seven synthetic peptides bearing different charges by means of Ion-Exchange chromatography. Reproduced with permission from [67].
Ion-exchange chromatography has been employed also for pre-purification of the peptide of interest. In a previous study, lactoferrin was hydrolyzed with porcin pepsin A. The hydrolysate was then loaded in a SP-Sepharose Fast Flow column (where SP is sulphopropyl, a strong cation exchanger) and eluted with a gradient of ammonia solution. During the gradient elution, only impurities were eluted. At the end of the gradient, a wash was performed with NaCl 2 M, in order to recover the target peptide (LFcin-B) [72]. Otherwise, it is possible to modulate the experimental conditions in order to trap the impurities on the resin whereas the peptide passes through the column with no retention. For example, this procedure was applied for the purification of C-peptide. This molecule contains 31 amino acid residues, with only one being basic, therefore it has a very low isoelectric point, around 3. By using a strong cation exchange and applying a mobile phase with pH slightly above its isolectric point, this strategy resulted to be successful, as it was confirmed by a comparison of LC-MS chromatograms of the sample before and after the purification [73].
Last, IEX is often employed for peptide mapping, to demonstrate protein identity, as in the case of cytochrome C tryptic digest and hemoglobin [65].
References
- Bhat, Z.F.; Kumar, S.; Bhat, H.F. Bioactive peptides of animal origin: A review. J. Food Sci. Technol. 2015, 52, 5377–5392.
- Möller, N.P.; Scholz-Ahrens, K.E.; Roos, N.; Schrezenmeir, J. Bioactive peptides and proteins from foods: Indication for health effects. Eur. J. Nutr. 2008, 47, 171–182.
- Fitzgerald, R.J.; Murray, B.A.; Walsh, D.J. Hypotensive peptides from milk proteins. J. Nutr. 2004, 134, 980–988.
- Gobbetti, M.; Minervini, F.; Rizzello, C.G. Bioactive peptides in diary products. In Handbook of Food Products Manufacturing; Wiley: Hoboken, NJ, USA, 2007.
- Korhonen, H. Milk-derived bioactive peptides: From science to applications. J. Funct. Food 2009, 1, 177–187.
- Meisel, H.; Fitzgerald, R.J. Biofunctional peptides from milk proteins: Mineral binding and cytomodulatory effects. Curr. Pharm. Des. 2003, 9, 1289–1295.
- Korhonen, H.; Pihlanto, A. Food-derived bioactive peptides-opportunities for designing future foods. Curr. Pharm. Des. 2003, 9, 1297–1308.
- Arihara, K.; Nakashima, Y.; Mukai, T.; Ishikawa, S.; Itoh, M. Peptide inhibitors for angiotensin I-converting enzyme from enzymatic hydrolysates of porcin skeletal muscle proteins. Meat Sci. 2001, 57, 319–324.
- Nakashima, Y.; Arihara, K.; Sasaki, A.; Ishikawa, S.; Itoh, M. Antihypertensive activities of peptides derived from porcin skeletal muscle myosin in spontaneously hypertensive rats. J. Food Sci. 2002, 67, 434–437.
- Murakami, Y.; Hirata, A. Novel process for enzymatic hydrolysis of proteins in an aqueous two-phase system for the production of peptide mixture. Prep. Biochem. Biotechnol. 2000, 30, 31–37.
- Martin-Orue, C.; Henry, G.; Bouhallab, S. Tryptic hydrolysis of k-caseino macropeptide: Control of the enzymatic reaction in a continuous membrane reactor. Enzym. Microb. Technol. 1999, 24, 173–180.
- Perea, A.; Ugalde, U. Continuous hydrolysis of whey proteins in a membrane recycle reactor. Enzym. Microb. Technol. 1996, 18, 29–34.
- Aihara, K.; Kajimoto, O.; Hirata, H.; Takahashi, R.; Nakamura, Y. Effect of powdered fermented milk with Lactobacillus helveticus on subjects with high normal blood pressure or mild hypertension. J. Am. Coll. Nutr. 2005, 24, 257–265.
- Chen, G.W.; Tsai, J.S.; Sun Pan, B. Purification of angiotensin I-converting enzyme inhibitory peptides and antihypertensive effect of milk produced by protease facilitated lactic fermentation. Int. Diary J. 2007, 17, 641–647.
- Hata, Y.; Yamamoto, M.; Ohni, H.; Nakajima, K.; Nakamura, Y.; Takano, T. A placebo-controlled study of the effect of sour milk on blood pressure in hypertensive subjects. Am. J. Clin. Nutr. 1996, 64, 767–771.
- Jauhiainen, T.; Vapaatalo, H.; Poussa, T.; Kyrönpalo, S.; Rasmussen, M.; Korpela, R. Lactobacillus helveticus fermented milk reduces blood pressure in 24-h ambulatory blood pressure measurements. Am. J. Hypertens. 2005, 18, 1600–1605.
- Masuda, O.; Nakamura, Y.; Takano, T. Antihypertensive peptides are present in aorta after oral administration of sourmilk containing these peptides to spontaneously hypertensive rats. J. Nutr. 1996, 126, 3063–3068.
- Seppo, L.; Jauhiainen, T.; Poussa, T.; Korpela, R. A fermented milk high in bioactive peptides has a blood pressure-lowering effect in hypertensive subjects. Am. J. Clin. Nutr. 2003, 77, 326–330.
- Saiga, A.; Okumura, T.; Makihara, T.; Katsuta, S.; Shimizu, T.; Yamada, R.; Nishimura, T. Angiotensin I-converting enzymes inhibitory peptides in a hydrolyzed chicken breast muscle extract. J. Agric. Food Chem. 2003, 51, 1740–1745.
- Becker, G.L. Preserving food and health: Antioxidants make functional, nutritious preservatives. Food Proc. 1993, 12, 54–56.
- Sakanaka, S.; Tachibana, Y.; Ishihara, N.; Juneja, L.R. Antioxidant properties of casein calcium peptides and their effects on lipid oxidation in beef homogenates. J. Agric. Food Chem. 2005, 53, 464–468.
- Albericio, F. Developments in peptide and amide synthesis. Curr. Opin. Chem. Biol. 2004, 4, 211–221.
- Merrifield, R.B. Solid Phase Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154.
- Wegmüller, S.; Schmid, S. Recombinant peptide production in microbial cells. Curr. Org. Chem. 2014, 18, 1005–1019.
- Guzmán, F.; Barberis, S.; Illantes, A. Peptide synthesis: Chemical or enzymatic. Electron. J. Biotechnol. 2007, 10, 279–314.
- Gill, I.; López-Fandiño, R.; Jorba, X.; Vulfson, E.N. Biologically active peptides and enzymatic approaches to their production. Enzym. Microb. Technol. 1996, 18, 162–183.
- De Luca, C.; Felletti, S.; Lievore, G.; Buratti, A.; Vogg, S.; Morbidelli, M.; Cavazzini, A.; Catani, M.; Macis, M.; Ricci, A.; et al. From batch to continuous chromatographic purification of a therapeutic peptide through Multicolumn Countercurrent Solvent Gradient Purification. J. Chromatogr. A 2020, 1625, 461304.
- De Luca, C.; Felletti, S.; Macis, M.; Cabri, W.; Lievore, G.; Chenet, T.; Pasti, L.; Morbidelli, M.; Cavazzini, A.; Catani, M.; et al. Modeling the nonlinear behavior of a bioactive peptide in reversed-phase gradient elution chromatography. J. Chromatogr. A 2020, 1616, 460789.
- Martelli, G.; Cantelmi, P.; Tolomelli, A.; Corbisiero, D.; Mattellone, A.; Ricci, A.; Fantoni, T.; Cabri, W.; Vacondio, F.; Ferlenghi, F.; et al. Steps towards sustainable solid phase peptide synthesis: Use and recovery of N-octyl pyrrolidone. Green Chem. 2021.
- Martelli, G.; Ferrazzano, L.; Tolomelli, A.; Ricci, A.; Cabri, W. Pharmaceutical Green Chemistry in Peptide Synthesi—A Snapshot on the Role of Solvents in SPPS. Chem. Today 2020, 38, 14–17.
- Ferrazzano, L.; Corbisiero, D.; Martelli, G.; Tolomelli, A.; Viola, A.; Ricci, A.; Cabri, W. Green Solvent Mixtures for Solid-Phase Peptide Synthesis: A Dimethylformamide-Free Highly Efficient Synthesis of Pharmaceutical-Grade Peptides. ACS Sustain. Chem. Eng. 2019, 7, 12867–12877.
- Bray, B.L. Large-scale manufacture of peptide therapeutics by chemical synthesis. Nat. Rev. Drug Discov. 2003, 2, 587–593.
- Li, Y. Recombinant production of antimicrobial peptides in Escherichia coli: A review. Protein Expr. Purif. 2011, 80, 260–267.
- Carta, G.; Jungbauer, A. Protein Chromatography. Process Development and Scale-Up; WILEY-VCH: Weinheim, Germany, 2010.
- Shaik, M.I.; Sarbon, N.M. A review on purification and characterization of anti-proliferative peptides derived from fish protein hydrolysate. Food Rev. Int. 2020.
- Turgeon, S.L.; Gauthier, S.F. Whey peptide fractions obtained with a two-step ultrafiltration process: Production and characterization. J. Food Sci. 1990, 55, 106–110.
- Farmerie, L.; Rustandi, R.R.; Loughney, J.W.; Dawod, M. Recent advances in isoelectric focusing of proteins and peptides. J. Chromatogr. A 2021, 1651, 462274.
- Ciborowski, P.; Silberring, J. Proteomic Profiling and Analytical Chemistry: The Crossroads; Elsevier: Amsterdam, The Netherlands, 2016.
- Šalplachta, J.; Horká, M.; Šlais, K. Preparative isoelectric focusing in a cellulose-based separation medium. J. Sep. Sci. 2017, 40, 2498–2505.
- Litowski, J.R.; Semchuk, P.D.; Mant, C.T.; Hodges, R.S. Hydrophilic interaction/cation-exchange chromatography for the purification of synthetic peptides from closely related impurities: Serine side-chain acetylated peptides. J. Pept. Res. 1999, 54, 1–11.
- Bodanszky, M. Undesired reactions during synthesis. In Peptide Chemistry; Springer: Berlin/Heidelberg, Germany, 1988; pp. 104–114.
- Erickson, B.W.; Merrifield, R.B. Solid-phase peptide synthesis. In The Proteins; Academic Press: Cambridge, MA, USA, 1976; pp. 397–432.
- Teoh, H.K.; Sorensen, E.; Titchener-Hooker, N. Optimal operating policies for for closed-loop recycling HPLC processes. Chem. Eng. Sci. 2003, 58, 4145–4158.
- Sainio, T.; Kaspereit, M. Analysis of steady-state recycling chromatography using equilibrium theory. Sep. Purif. Technol. 2009, 66, 9–18.
- Agyei, D.; Ongkudon, C.M.; Wei, C.Y.; Chan, A.S.; Danquah, M.K. Bioprocess challenges to the isolation and purification of bioactive peptides. Food Bioprod. Proc. 2016, 98, 244–256.
- Makey, D.M.; Shchurik, V.; Wang, H.; Lhotka, H.R.; Stoll, D.R.; Vazhentsev, A.; Mangion, I.; Regalado, E.L.; Haidar Ahmad, I.A. Mapping the Separation Landscape in Two-Dimensional Liquid Chromatography: Blueprints for Efficient Analysis and Purification of Pharmaceuticals Enabled by Computer-Assisted Modeling. Anal. Chem. 2021, 93, 964–972.
- Khalaf, R.; Forrer, N.; Buffolino, G.; Gétaz, D.; Bernardi, S.; Butté, A.; Morbidelli, M. Doping reversed-phase media for improved peptide purification. J. Chromatogr. A 2015, 1397, 11–18.
- Zhang, X.; Fang, A.; Riley, C.P.; Wang, M.; Regnier, F.E.; Buck, C. Multi-dimensional liquid chromatography in proteomics—A review. Anal. Chim. Acta 2010, 664, 101–113.
- D’Attoma, A.; Heinisch, S. On-line comprehensive two dimensional separations of charged compounds using reversed-phase high performance liquid chromatography and hydrophilic interaction chromatography. Part II: Application to the separation of peptides. J. Chromatogr. A 2013, 1306, 27–36.
- Zhang, Y.; Zeng, L.; Pham, C.; Xu, R. Preparative two-dimensional liquid chromatography/mass spectrometry for the purification of complex pharmaceutical samples. J. Chromatogr. A 2014, 1324, 86–95.
- Clarke, A.; Ehkirch, A. The Current Status and Future of Two- and Multidimensional Liquid Chromatography in Pharmaceutical R&D and QC. LC-GC Eur. 2020, 33, 136–150.
- Stoll, D.R.; Carr, P.W. Two-Dimensional Liquid Chromatography: A State of the Art Tutorial. Anal. Chem. 2017, 89, 519–531.
- Åsberg, D.; Langborg Weinmann, A.; Leek, T.; Lewis, R.J.; Klarqvist, M.; Leśko, M.; Kaczmarski, K.; Samuelsson, J.; Fornstedt, T. The importance of ion-pairing in peptide purification by reversed-phase liquid chromatography. J. Chromatogr. A 2017, 1496, 80–91.
- Toll, H.; Oberacher, H.; Swart, R.; Huber, C.G. Separation, detection and identification of peptides by ion-pair reversed-phase high-performance liquid chromatography-electrospray ionization mass spectrometry at high and low pH. J. Chromatogr. A 2005, 1079, 274–286.
- Ruta, J.; Guillarme, D.; Rudaz, S.; Veuthey, J.L. Comparison of columns packed with porous sub-2 µm particles and superficially porous sub-3 µm particles for peptide analysis at ambient and high temperature. J. Sep. Sci. 2010, 33, 2465–2477.
- De Luca, C.; Felletti, S.; Lievore, G.; Buratti, A.; Chenet, T.; Pasti, L.; Morbidelli, M.; Cavazzini, A.; Catani, M.; Macis, M.; et al. Determination of the thermodynamic behavior of a therapeutic peptide in overloading conditions in gradient elution chromatography. J. Chromatogr. Sep. Technol. 2020, 11, 428.
- Moslemi, P.; Najafabadi, A.R.; Tajerzadeh, H. Rapid and sensitive method for simultaneous determination of insulin and A21-desamido insulin by high-performance liquid chromatography. J. Pharm. Biomed. Anal. 2003, 33, 45–51.
- Harris, P.W.R.; Lee, D.J.; Brimble, M.A. A slow gradient approach for the purification of synthetic polypeptides by reversed phase high performance liquid chromatography. J. Pept. Sci. 2012, 18, 549–555.
- Brettschneider, F.; Jankowski, V.; Günthner, T.; Salem, S.; Nierhaus, M.; Schulz, A.; Zidek, W.; Jankowski, J. Replacement of acetonitrile by ethanol as solvent in reversed phase chromatography of biomolecules. J. Chromatogr. B 2010, 878, 763–768.
- Howard, J.W.; Kay, R.G.; Pleasance, S.; Creaser, C.S. UHPLC for the separation of proteins and peptides. Bioanalysis 2012, 4, 2971–2988.
- Tarafder, A.; Aumann, L.; Morbidelli, M. The role of ion-pairing in peak deformations in overloaded reversed-phase chromatography of peptides. J. Chromatogr. A 2010, 1217, 7065–7073.
- Bobály, B.; Mikola, V.; Sipkó, E.; Márta, Z.; Fekete, J. Recovery of proteins affected by mobile phase trifluoroacetic acid concentration in reversed-phase chromatography. J. Chromatogr. Sci. 2015, 53, 1078–1083.
- Mazzoccanti, G.; Manetto, S.; Bassan, M.; Foschini, A.; Orlandin, A.; Ricci, A.; Cabri, W.; Ismail, O.H.; Catani, M.; Cavazzini, A.; et al. Boosting basic-peptide separation through dynamic electrostatic-repulsion reversed-phase (d-ERRP) liquid chromatography. RSC Adv. 2020, 10, 12604–12610.
- Fekete, S.; Veuthey, J.L.; Guillarme, D. New trends in reversed-phase liquid chromatographic separations of therapeutic peptides and proteins: Theory and applications. J. Pharm. Biomed. Anal. 2012, 69, 9–27.
- Fekete, S.; Beck, A.; Veuthey, J.L.; Guillarme, D. Ion-exchange chromatography for the characterization of biopharmaceuticals. J. Pharm. Biomed. Anal. 2015, 113, 43–55.
- Waite, S.; McGinley, M. Peptide Separations by Cation Exchange Chromatography Using Luna SCX; Application Note TN-1024; Phenomenex Inc.: Torrance, CA, USA, 2005.
- Crimmins, D.L. Strong cation-exchange high-performance liquid chromatography as a versatile tool for the characterization and purification of peptides. Anal. Chim. Acta 1997, 352, 21–30.
- Alpert, A.J. Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compound. J. Chromatogr. A 1990, 499, 177–196.
- Alpert, A.J.; Andrews, P.C. Cation-exchange chromatography of peptides on poly(2-sulfoethyl aspartamide)-silica. J. Chromatogr. A 1988, 443, 85–96.
- Crimmins, D.L.; Gorka, J.; Thoma, R.S.; Schwartz, B.D. Peptide characterization with a sulfoethyl aspartamide column. J. Chromatogr. A 1988, 443, 63–71.
- Crimmins, D.L.; Thoma, R.S.; McCourt, D.W.; Schwartz, B.D. Strong-cation-exchange sulfoethyl aspartamide chromatography for peptide mapping of Staphylococcus aureus V8 protein digests. Anal. Biochem. 1989, 176, 255–260.
- Recio, I.; Visser, S. Two ion-exchange chromatographic methods for the isolation of antibacterial peptides from lactoferrin: In situ enzymatic hydrolysis on an ion-exchange membrane. J. Chromatogr. A 1999, 831, 191–201.
- Stoyanov, A.V.; Rohlfing, C.L.; Connolly, S.; Roberts, M.L.; Nauser, C.L.; Little, R.R. Use of cation exchange chromatography for human C-peptide isotope dilution—Mass spectrometric assay. J. Chromatogr. A 2011, 1218, 9244–9249.

