 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Varun Kumar | + 1698 word(s) | 1698 | 2021-08-05 11:58:16 | | | |
| 2 | Lindsay Dong | Meta information modification | 1698 | 2021-08-27 03:15:36 | | |
Video Upload Options
Fuchs endothelial corneal dystrophy (FECD) is a genetically complex, heterogenous, age-related degenerative disease of corneal endothelial cells (CEnCs), occurring in the fifth decade of life with a higher incidence in females. It is characterized by extracellular matrix (ECM) protein deposition called corneal guttae, causing light glare and visual complaints in patients. In FECD, CEnCs exhibit stress-induced senescence, oxidative stress, DNA damage, heightened reactive oxygen species (ROS) production, mitochondrial damage, and dysfunction as well as sustained endoplasmic reticulum (ER) stress. Among all of these, mitochondrial dysfunction involving altered mitochondrial bioenergetics and dynamics plays a critical role in FECD pathogenesis.
1. Introduction
The corneal endothelial is the innermost layer of the cornea and plays an important role in maintaining water balance and clarity of the cornea. Fuchs endothelial corneal dystrophy (FECD) is the most common corneal endothelial dystrophy. It is a bilateral, genetically heterogeneous degenerative disease of CEnCs occurring in 4% of the U.S. population over 40 years of age, with a higher incidence in women [1][2][3][4]. It is characterized by the progressive decline of the CEnCs and the formation of extracellular matrix excrescences [5][6][7] in Descemet’s membrane (DM), called guttae, leading to corneal edema and loss of vision. Currently, the only treatment for FECD is corneal transplantation, which accounts for approximately 32,000 of the endothelial keratoplasties performed in the U.S. annually. It carries substantial economic and social burdens. Understanding the disease pathogenesis is essential for developing pharmacotherapeutic interventions to halt the disease. In FECD, CEnCs exhibits stress-induced senescence [8], oxidant-antioxidant imbalance [9], mitochondrial DNA damage [10] and dysfunction [11], sustained unfolded protein response (UPR) [12][13], and endoplasmic reticulum (ER) stress [14]. Among these factors, mitochondrial stress [15][16][17][18] plays an important role in FECD pathogenesis. Maintaining functional mitochondria is the key to the ion pump function in CEnCs. Excessive damage to the mitochondria leads to its selective degradation called auto(mito)phagy [15][18].
2. Mitochondria in CEnCs
Due to many ion pumps and very active endothelial cell metabolism, mitochondrial density is very high in CEnCs, second only to retinal photoreceptors [19]. Mitochondria, the powerhouse of the cells, regulate many physiological processes in CEnCs and play a pivotal role in their survival [20][21][22].
3. Mitochondrial DNA Damage in FECD
Mitochondrial DNA (mtDNA) damage occurs in many neurodegenerative disorders [23] such as Alzheimer’s, Parkinson’s, and Huntington’s diseases. It also occurs in retinal diseases [24] such as age-related macular degeneration, diabetic retinopathy, glaucoma, and in corneal diseases [25] such as keratoconus, Kearns Sayre Syndrome, and FECD [26][27]. In human FECD tissues, we found that 8-hydroxydeoxyguanosine (8-OHdG), a marker of oxidized DNA lesions, accumulated in mtDNA of CEnCs, suggesting increased oxidative mtDNA damage [9]. Further studies using long-amplicon-quantitative polymerase chain reaction (LA-qPCR) demonstrated that human FECD specimens had significantly decreased small mitochondrial copy number and increased DNA lesion frequency (indicative of damage) than the normal specimens [11].
As Fuchs is prevalent in females [2][3], Liu et al. analyzed mtDNA damage in the mouse model of ultraviolet A (UVA)-induced FECD for both sexes and found that mtDNA damage was significantly more in the mouse corneal endothelial cells (MCEnCs) at week 4 and 8 post-UVA in females compared to males, suggesting the female susceptibility to FECD [10]. However, mtDNA exhibited a similar extent of damage in both male and female mice at day 1 post-UVA, and it recovered at week 2 post-UVA [10].
For the in vitro studies, menadione (MN) induced oxidative stress to study mtDNA damage in normal (HCEnC-21T or HCECi: normal telomerase immortalized human corneal endothelial) and Fuchs (FECDi: immortalized FECD cell lines derived from FECD specimens) cell lines. Halilovic et al. demonstrated that the FECD cell line had significantly reduced the small mtDNA copy number compared to the normal control cell line and remained low in quantity after MN exposure [11]. MtDNA damage was significantly greater in the control cell lines than the untreated control and remained significantly high in the FECD cell line after MN exposure [11]. Miyajima et al. demonstrated a dysfunctional Nrf2-NQO1 axis, and specifically loss of NQO1 (NAD(P)H:quinone oxidoreductase 1) protein in FECD contributes to mitochondrial DNA damage and estrogen genotoxicity, explaining the higher incidence of FECD in females [28].
4. Mitochondrial Dysfunction in FECD
5. Autophagy and Mitophagy
6. Mechanisms of Mitophagy in FECD
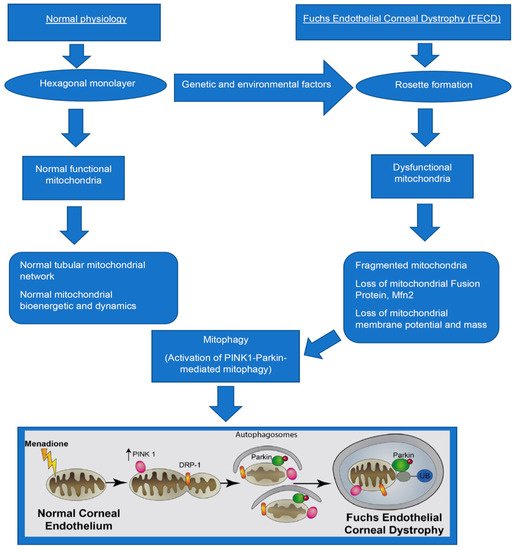
7. Role of Mitophagy in FECD
The activation of mitophagy being detrimental or useful remains unclear in FECD. PINK1-Parkin mediated mitophagy, as described by our group, suggests that excessive mitophagy might destroy many normal mitochondrial, disturbing mitochondrial bioenergetics, and contributing to the cascade of destructive events in the pathogenesis of FECD. Therefore, therapeutics targeting mitophagy might not be beneficial in the late stage of FECD. However, it might be advantageous in the earlier stages of FECD as CEnCs might alleviate some intracellular mitochondrial stress by removing damaged mitochondria via mitophagy.
References
- Zhang, C.; Bell, W.R.; Sundin, O.H.; de La Cruz, Z.; Stark, W.J.; Green, W.R.; Gottsch, J.D. Immunohistochemistry and electron microscopy of early-onset Fuchs corneal dystrophy in three cases with the same L450W COL8A2 mutation. Trans. Am. Oph-Thalmol. Soc. 2006, 104, 85–97.
- Krachmer, J.H.; Purcell, J.J., Jr.; Young, C.W.; Bucher, K.D. Corneal endothelial dystrophy. A study of 64 families. Arch. Ophthalmol. 1978, 96, 2036–2039.
- Rosenblum, P.; Stark, W.J.; Maumenee, I.H.; Hirst, L.W.; Maumenee, A.E. Hereditary Fuchs’ dystrophy. Am. J. Ophthalmol. 1980, 90, 455–462.
- Bahn, C.F.; Falls, H.F.; Varley, G.A.; Meyer, R.F.; Edelhauser, H.F.; Bourne, W.M. Classification of corneal endothelial disorders based on neural crest origin. Ophthalmology 1984, 91, 558–563.
- Goyer, B.; Thériault, M.; Gendron, S.P.; Brunette, I.; Rochette, P.; Proulx, S. Extracellular matrix and integrin expression profiles in Fuchs endothelial corneal dystrophy cells and tissue model. Tissue Eng. Part A 2018, 24, 607–615.
- Weller, J.M.; Zenkel, M.; Schlötzer-Schrehardt, U.; Bachmann, B.O.; Tourtas, T.; Kruse, F.E. Extracellular matrix alterations in late-onset Fuchs’ corneal dystrophy. Investig. Opthalmol. Vis. Sci. 2014, 55, 3700–3708.
- Poulsen, E.T.; Dyrlund, T.F.; Runager, K.; Scavenius, C.; Krogager, T.P.; Højrup, P.; Thøgersen, I.B.; Sanggaard, K.W.; Vorum, H.; Hjortdal, J.; et al. Proteomics of Fuchs’ endothelial corneal dystrophy support that the extracellular matrix of descemet’s membrane is disordered. J. Proteome Res. 2014, 13, 4659–4667.
- Matthaei, M.; Meng, H.; Meeker, A.K.; Eberhart, C.G.; Jun, A.S. Endothelial Cdkn1a (p21) overexpression and accelerated se-nescence in a mouse model of Fuchs endothelial corneal dystrophy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6718–6727.
- Jurkunas, U.V.; Bitar, M.S.; Funaki, T.; Azizi, B. Evidence of oxidative stress in the pathogenesis of Fuchs endothelial corneal dystrophy. Am. J. Pathol. 2010, 177, 2278–2289.
- Liu, C.; Miyajima, T.; Melangath, G.; Miyai, T.; Vasanth, S.; Deshpande, N.; Kumar, V.; Tone, S.O.; Gupta, R.; Zhu, S.; et al. Ultraviolet A light induces DNA damage and estrogen-DNA adducts in Fuchs endothelial corneal dystrophy causing females to be more affected. Proc. Natl. Acad. Sci. USA 2020, 117, 573–583.
- Halilovic, A.; Schmedt, T.; Benischke, A.-S.; Hamill, C.; Chen, Y.; Santos, J.H.; Jurkunas, U.V. Menadione-induced DNA damage leads to mitochondrial dysfunction and fragmentation during rosette formation in Fuchs endothelial corneal dystrophy. Antioxid. Redox Signal. 2016, 24, 1072–1083.
- Engler, C.; Kelliher, C.; Spitze, A.R.; Speck, C.L.; Eberhart, C.G.; Jun, A.S. Unfolded protein response in Fuchs endothelial corneal dystrophy: A unifying pathogenic pathway? Am. J. Ophthalmol. 2010, 149, 194–202.e2.
- Okumura, N.; Hashimoto, K.; Kitahara, M.; Okuda, H.; Ueda, E.; Watanabe, K.; Nakahara, M.; Sato, T.; Kinoshita, S.; Tourtas, T.; et al. Activation of TGF-beta signaling induces cell death via the unfolded protein response in Fuchs endothelial corneal dys-trophy. Sci. Rep. 2017, 7, 6801.
- Okumura, N.; Kitahara, M.; Okuda, H.; Hashimoto, K.; Ueda, E.; Nakahara, M.; Kinoshita, S.; Young, R.D.; Quantock, A.J.; Tourtas, T.; et al. Sustained activation of the unfolded protein response induces cell death in Fuchs’ endothelial corneal dystrophy. Investig. Opthalmol. Vis. Sci. 2017, 58, 3697–3707.
- Benischke, A.S.; Vasanth, S.; Miyai, T.; Katikireddy, K.R.; White, T.; Chen, Y.; Halilovic, A.; Price, M.; Price, F., Jr.; Liton, P.B.; et al. Activation of mitophagy leads to decline in Mfn2 and loss of mitochondrial mass in Fuchs endothelial corneal dystrophy. Sci. Rep. 2017, 7, 6656.
- Méthot, S.J.; Proulx, S.; Brunette, I.; Rochette, P.J. Chronology of cellular events related to mitochondrial burnout leading to cell death in Fuchs endothelial corneal dystrophy. Sci. Rep. 2020, 10, 1–10.
- Li, Y.J.; Minear, M.A.; Qin, X.; Rimmler, J.; Hauser, M.A.; Allingham, R.R.; Igo, R.P.; Lass, J.H.; Iyengar, S.K.; Klintworth, G.K.; et al. Mitochondrial polymorphism A10398G and Haplogroup I are associated with Fuchs’ endothelial corneal dystrophy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4577–4584.
- Miyai, T.; Vasanth, S.; Melangath, G.; Deshpande, N.; Kumar, V.; Benischke, A.-S.; Chen, Y.; Price, M.O.; Price, F.W.; Jurkunas, U.V. Activation of PINK1-parkin–mediated mitophagy degrades mitochondrial quality control proteins in Fuchs endothelial corneal dystrophy. Am. J. Pathol. 2019, 189, 2061–2076.
- Hogan, M.J. Histology of the Human Eye: An Atlas and Textbook; Saunders: Philadelphia, PA, USA, 1971.
- Suomalainen, A.; Battersby, B.J. Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 2018, 19, 77–92.
- Tzameli, I. The evolving role of mitochondria in metabolism. Trends Endocrinol. Metab. 2012, 23, 417–419.
- Tait, S.W.; Green, D.R. Mitochondrial regulation of cell death. Cold Spring Harb. Perspect. Biol. 2013, 5, a008706.
- Yang, J.-L.; Weissman, L.; Bohr, V.A.; Mattson, M.P. Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair 2008, 7, 1110–1120.
- Jarrett, S.; Lin, H.; Godley, B.F.; Boulton, M.E. Mitochondrial DNA damage and its potential role in retinal degeneration. Prog. Retin. Eye Res. 2008, 27, 596–607.
- Vallabh, N.A.; Romano, V.; Willoughby, C.E. Mitochondrial dysfunction and oxidative stress in corneal disease. Mitochondrion 2017, 36, 103–113.
- Czarny, P.; Seda, A.; Wielgorski, M.; Binczyk, E.; Markiewicz, B.; Kasprzak, E.; Jimenez-Garcia, M.P.; Grabska-Liberek, I.; Pawlowska, E.; Blasiak, J.; et al. Mutagenesis of mitochondrial DNA in Fuchs endothelial corneal dystrophy. Mutat. Res. Mol. Mech. Mutagen. 2014, 760, 42–47.
- Gendron, S.P.; Thériault, M.; Proulx, S.; Brunette, I.; Rochette, P.J. Restoration of mitochondrial integrity, telomere length, and sensitivity to oxidation by in vitro culture of Fuchs’ endothelial corneal dystrophy cells. Investig. Opthalmol. Vis. Sci. 2016, 57, 5926–5934.
- Miyajima, T.; Melangath, G.; Zhu, S.; Deshpande, N.; Vasanth, S.; Mondal, B.; Kumar, V.; Chen, Y.; Price, M.O.; Price, F.W.; et al. Loss of NQO1 generates genotoxic estrogen-DNA adducts in Fuchs endothelial corneal dystrophy. Free Radic. Biol. Med. 2020, 147, 69–79.
- Hu, F.; Liu, F. Mitochondrial stress: A bridge between mitochondrial dysfunction and metabolic diseases? Cell. Signal. 2011, 23, 1528–1533.
- Hill, S.; Sataranatarajan, K.; van Remmen, H. Role of signaling molecules in mitochondrial stress response. Front. Genet. 2018, 9, 225.
- Hill, S.; van Remmen, H. Mitochondrial stress signaling in longevity: A new role for mitochondrial function in aging. Redox Biol. 2014, 2, 936–944.
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014.
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280.
- Sifuentes-Franco, S.; Padilla-Tejeda, D.E.; Carrillo-Ibarra, S.; Miranda-Díaz, A.G. Oxidative Stress, Apoptosis, and Mitochondrial Function in Diabetic Nephropathy. Int. J. Endocrinol. 2018, 2018, 1–13.
- Payne, B.A.I.; Chinnery, P.F. Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1347–1353.
- Calvani, R.; Joseph, A.-M.; Adhihetty, P.J.; Miccheli, A.; Bossola, M.; Leeuwenburgh, C.; Bernabei, R.; Marzetti, E. Mitochondrial pathways in sarcopenia of aging and disuse muscle atrophy. Biol. Chem. 2013, 394, 393–414.
- Borboli, S.; Colby, K. Mechanisms of disease: Fuchs’ endothelial dystrophy. Ophthalmol. Clin. N. Am. 2002, 15, 17–25.
- Tuberville, A.W.; Wood, T.O.; McLaughlin, B.J. Cytochrome oxidase activity of Fuchs’ endothelial dystrophy. Curr. Eye Res. 1986, 5, 939–947.
- Meng, H.; Matthaei, M.; Ramanan, N.; Grebe, R.; Chakravarti, S.; Speck, C.L.; Kimos, M.; Vij, N.; Eberhart, C.G.; Jun, A.S. L450W and Q455K Col8a2 knock-in mouse models of Fuchs endothelial corneal dystrophy show distinct phenotypes and evidence for altered autophagy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1887–1897.


