 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nina Kunová | + 3198 word(s) | 3198 | 2021-08-03 11:11:47 | | | |
| 2 | Ron Wang | + 299 word(s) | 3497 | 2021-08-26 11:23:13 | | | | |
| 3 | Ron Wang | + 299 word(s) | 3497 | 2021-08-26 11:35:08 | | |
Video Upload Options
Mitochondrial HSP70 (mtHSP70/GRP75/HSPA9/PBP74), also called mortalin, is an essential protein belonging to the HSP70 sub-family which has great importance for mitochondrial biogenesis and the correct functioning of the whole cellular machinery.
1. Introduction
The mitochondrial protein quality control (PQC) system is a network-like organization of chaperones and proteases whose major role is to preserve the functional and active states of mitochondrial proteins under diverse, and sometimes pathogenic, conditions. PQC maintains protein homeostasis or proteostasis among the mitochondrial proteins by controlling the balance between the generation of newly synthesized proteins and the removal of damaged or misfolded proteins beyond the scope of repair or refolding.
Protein damage, in the context of its structure and functionality, usually means a loss of function combined with changes to its native conformational state, both of which might occur as a result of non-physiological temperatures (heat stress) or chemical modifications (e.g., reactive oxygen species (ROS)) causing oxidative stress, which in humans may lead to a wide variety of pathologies, including cancer, neurodegenerative disorders, diabetes, cardiovascular diseases, atherosclerosis, stroke, inflammatory disorders, chronic fatigue syndrome, asthma, and age-related pathologies (review in [1]). Misfolded proteins are prone to form abnormal or irregular interactions as a result of the exposure of their normally buried hydrophobic parts; this often leads to the toxic accumulation of insoluble aggregates [2]. Interestingly, even a slight increase in temperature can trigger a heat shock response. The problem is not the temperature itself, but the protein unfolding, entanglement, and non-specific aggregation that the heat causes. In eukaryotes, the major damage appears to consist of disruption to the cytoskeleton (reorganization of actin filaments and tubulin networks), fragmentation of the membranous organelles (the Golgi complex and endoplasmic reticulum), aggregation of ribosomal proteins, and a decrease in the number of mitochondria and lysosomes [3]. Mitochondria loss leads to a dramatic drop in oxidative phosphorylation and ATP levels, causing a further imbalance in homeostasis, ultimately leading to cell death [4]. To avoid this fate, cells possess so-called heat shock proteins (HSPs), whose expression increases in response to such stresses, and which serve as the foundation of resistance to hostile conditions [3].
HSPs primarily act as molecular chaperones that monitor and regulate polypeptide folding and prevent the aggregation or precipitation of misfolded proteins. They are often overexpressed upon stress, in situations that would normally be lethal. Oxidative, cytokine and muscular stresses; nutritional deficiencies; viral infections; hyperthermia; ischemia and alterations in calcium and pH in different types of cells and tissues are all potent inducers of increased HSP levels. In humans, their deregulation often underlies the pathologies of several diseases, including such devastating neurological disorders as Alzheimer’s disease (AD), Parkinson’s disease (PD), Creutzfeldt-Jacobs disease, Huntington’s disease, prion-related diseases, and amyotrophic lateral sclerosis (ALS) [5][6].
The accumulation of misfolded proteins and proteins irreversibly modified by post-translational modifications (e.g., protein glycation, methionine oxidation, deamination of asparaginyl and glutaminyl residues) that induce conformational changes and impaired protein functions has also been observed in ageing [5]. Such alterations cannot be simply reversed by molecular chaperones, these may only accompany their substrates and by a stable association with their hydrophobic surfaces prevent their aggregation thus shifting the normal functions of HSPs from protein maintenance to fighting the growing number of damaged or non-functional enzymes [5][7]. The vulnerability of proteins to aggregation poses a great danger and the only solution in protecting the cell itself is their complete removal. However, ageing also decreases the activity of the proteasome, the main cytosolic proteolytic enzyme [8][9]. Therefore, some species developed constitutively induced chaperones (small heat shock proteins (sHSPs) and HSC70), which could fill the roles of impaired HSPs in ageing cells [10]. Under normal conditions, cytosolic HSC70 shuttles between the cytoplasm and the nucleus [11], which secures an export of nuclear proteins targeted for degradation [12]. The shuttling is transiently inhibited upon stress, when HSC70 becomes sequestered within the nucleus [13], thus protecting the stressed cells against possible damage and ensuring their survival when conditions improve [14]. When cells are exposed to excessive stress, a common response is to undergo cell death by either necrosis or apoptosis. HSPs are responsible for inhibiting both apoptotic and necrotic pathways and thus, they are also involved in cancer progression. The action of HSPs may cause uncontrolled cell growth, reduced tumor suppression, enhanced cell survival and may fuel tumor cell invasion, metastasis, and angiogenesis [15]. In various human cancers, HSPs were found to be expressed at high levels, providing an environment for tumor development and leading to poor patient prognosis and a resistance to therapy. Therefore, HSPs could also serve as biomarkers of cancer formation in several tissues and show the degree of progression and aggression of several types of tumors [16].
2. The 70-kDa Heat Shock Proteins (HSP70s)
Of all molecular chaperones, the HSP70 sub-family occupies a central position in every cellular proteostatic activity, from protein folding to disaggregation and degradation [17][18]. Its representatives are found in archaebacteria, prokaryotes, and eukaryotes, including plants and mammals [19], and possess one of the highest levels of conservation of all organismal proteins, with around 40–60% identity between the prokaryotic and eukaryotic homologues [20][21].
HSP70s reside in the cytoplasm of prokaryotes and in all major eukaryotic cellular sub-compartments [22][23][24]. At least one HSP70-encoding gene is expressed in all prokaryotes and eukaryotes. For example, E. coli harbors three Hsp70 isoforms: in addition to DnaK, the most thoroughly studied one, which mainly controls the folding of newly synthesized cytosolic proteins, two other HSP70 proteins have been identified, HscA and HscC [25][26]. HscA works together with its co-chaperone HscB in a chaperone/co-chaperone pair similar to that of DnaK/DnaJ [27][28]. It is required for the maturation of iron-sulfur clusters (ISC) [ 85,86] and is induced by cold stress [29]. HscC is heat induced, possesses ATPase activity, and is likely a chaperone [30], but does not act against denatured proteins [31]. S. cerevisiae expresses in total 11 HSP70 paralogues: 4 semi-redundant cytosolic/nuclear forms (SSA1, SSA2, SSA3, SSA4), 3 ribosome-associated chaperones (SSB1, SSB2, SSZ1), 3 mitochondrial chaperones (SSC1, SSQ1, SSC3) and 1 form specific for the endoplasmic reticulum (KAR2) [19][25][32][33]. Humans express 13 HSP70 homologues in different cellular compartments, including the cytosol and nucleus (HSPA1A/B, HSPA1L, HSPA2, HSPA6, HSPA7, HSPA8, HSPA12A/B, HSPA13, HSPA14), the ER (HSPA5) and the mitochondria (HSPA9) [25]. Human HSP70s differ not only in cellular localization but also in activity and expression [33][34]. Despite the large number of human isoforms, HSPA8 (also called HSC70) is the major, non-inducible cytosolic HSP70. It is constitutively expressed and provides the essential housekeeping functions in cellular protein quality control. Recent findings have also emphasized its involvement in regulating lysosome activity in specialized autophagy pathway called chaperone-mediated autophagy (CMA) (review in [35]). The second most abundant cytosolic homologue is the stress-inducible form of HSP70, HSPA1A (also named HSP72), whose expression increases in response to the accumulation of damaged or misfolded proteins [19][33][36].
In general, HSP70s consist of an N-terminal nucleotide-binding domain (NBD) and a C-terminal substrate-binding domain (SBD) connected by a flexible and highly conserved hydrophobic linker, which is crucial for allosteric inter-domain communication (review in [25]). The NBD domain is formed of four subdomains (Ia, IIa, Ib and IIb), organized into two lobes separated by a deep cleft, where the ATPase catalytic site resides ( Figure 1 a). In E. coli , the Ia NBD subdomain functions as a key mediator of inter-domain allostery, representing a signal transduction between the binding sites for ATP and substrate [37]. The SBD domain is capable of binding extended polypeptides rich in aliphatic residues and is composed of two parts, a β-sandwich subdomain (SBDβ) with the binding site and an α-helical subdomain (SBDα) acting as its flexible lid [25][36][38][39][40]. Both domains, NBD and SBD, are linked by a flexible linker that transfers the structural re-arrangements caused by the ATP hydrolysis from NBD to the SBD, which enables folding of the client protein [25].
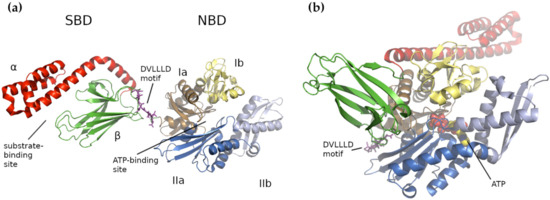
3. Human Mitochondrial HSP70 (mtHSP70)
Mitochondrial HSP70 (mtHSP70/GRP75/HSPA9/PBP74), also called mortalin, is an essential protein belonging to the HSP70 sub-family which has great importance for mitochondrial biogenesis and the correct functioning of the whole cellular machinery [52].
Mortalin was first identified in cell fusion studies of normal and immortal mouse fibroblasts as a marker of the mortal or lethal phenotype [53]. Mortalin was classified as a HSP70 stress chaperone based on its high degree of homology to other HSP70 members, including E. coli DnaK (51%), S. cerevisiae SSC1 (65%), and the rat cytosolic HSP70, HSC70 (46%) [53]. In humans, mortalin is a 74 kDa (679 amino acid), constitutively expressed protein and is one of the most abundant proteins in the mitochondrial matrix, accounting for approximately 1% of its total protein content [54]. Although mortalin is predominantly found in the mitochondrial matrix, when overexpressed it can also be found in extramitochondrial sites, including the cytosol and the perinuclear region.
Mortalin has the canonical HSP70 family structure, with a ~42 kDa NBD and a ~25 kDa PBD connected by a short hydrophobic linker (D434VLLLDVTP442), used for allosteric regulation by its co-chaperones [55][56][57]. Since mortalin is predominantly a mitochondrial protein, it also has a 46 residue mitochondrial pre-sequence at its N-terminus [58][59][60], but its C-terminus has the sequence K671EDQKEEKQ679, which differs from the EEVD motif found in other human HSP70 homologues [61].
In mitochondria, mortalin performs two specific roles: as a chaperone and stress-survival factor, it assists in protein quality control by (re)folding or degrading non-functional proteins [62][63][64], and as an essential component of the presequence translocase-associated motor (PAM), it binds precursor proteins to promote their unidirectional [65][62][66][67] translocation across the two mitochondrial membranes and into the mitochondrial matrix [67]. An excellent review by Pfanner et al. [68] provides a more detailed characterization of the biogenesis of mitochondrial proteins. In addition to mortalin, the PAM complex contains TOM20 and TOM22, the receptors on the surface of the mitochondrial outer membrane which recognize the cytosolic precursors carrying the mitochondrial localization signal (these form positively-charged amphipathic α-helices) [69][70]. These cytosolic pre-proteins are subsequently transported through the major outer membrane protein translocation channel, TOM40, and are then engaged by the presequence translocase of the inner membrane, TIM23 [71][72][73]. In the PAM complex, mortalin is the central ATP-driven chaperone and, together with its co-chaperones, ensures the translocation of the pre-protein into the mitochondrial matrix [68]. In humans, mortalin was shown to directly interact with the TIM23 complex, as well as cardiolipin-containing lipid bilayers, enabling its insertion into the inner mitochondrial membrane [74]. Although mortalin is approximately ten times more abundant than TIM23 [68], only a small amount of it acts in the TIM23-associated PAM in driving pre-protein import; the majority is involved in mitochondrial protein folding [73]. The overall maintenance of mitochondrial homeostasis is ensured by the cooperation of mortalin with the HSP60–HSP10 chaperonin complex; together, these play a central role in the correct folding of matrix-localized proteins, preventing protein misfolding and promoting the refolding and proper assembly of unfolded polypeptides that are generated under stress conditions inside the mitochondria [75].
In addition, mortalin also plays an important role in iron-sulfur cluster (ISC) biogenesis within the matrix and the proper insertion of Fe-S apoproteins [76][77][78]. It also closely cooperates with other mitochondrial homeostasis maintenance factors, including the tumor necrosis factor receptor-associated protein type 1 (TRAP-1) [79][80] and the mitochondrial voltage-dependent anion channel (VDAC) [81]. It regulates mitochondrial properties, including ATP levels, membrane potential and permeability [82][83], and it associates with the mitochondrial contact site and cristae organizing system (MICOS) complex [84]. Moreover, together with HSP60 and the mitochondrial ATP-dependent protease LON, mortalin forms part of the peripheral region of human mitochondrial nucleoids [85]. Outside the mitochondria, mortalin is also involved in other cellular activities, such as regulation of p53 activity, calcium and ROS signaling, intracellular trafficking, control of centrosome duplication, differentiation, and many others [86][87][88][89][90][91].
4. Human mtHSP70 Co-Chaperones
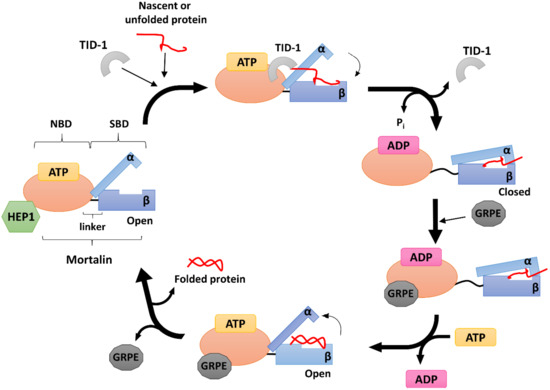
4.1 HEP1
The mortalin escort protein HEP1 (also called Zim17/TIM15/DNLZ) is a small (15 kDa) DNL‐type zinc finger protein that functions as a mtHSP70 partner on the matrix side of the inner mitochondrial membrane (Figure 2). Structurally, HEP1 forms asymmetric monomers, but it can also oligomerize depending on its actual concentration with Trp115 playing an important role in the process. Trp115 as the part of the zinc-binding domain (ZDB), lies on the surface of the monomer. The zinc finger motif seems particularly important for HEP1 itself, as well as for its interaction with mtHSP70. It has been shown that HEP1 is highly unstable in the presence of the EDTA chelator, indicating that the zinc ions are crucial for stabilizing its structure [92]. Moreover, when deletions or mutations in this motif occurs, HEP1 loses the ability to bind mortalin [93].
Studies in yeast demonstrated that HEP1 is essential for the mitochondrial import machinery. Deletion of its gene impaired the TIM23-dependent import of mitochondrial pre-proteins, which are necessary for yeast growth at elevated temperatures. When complexed with mtHSP70, HEP1 has a dual character: it protects mtHSP70 from self-aggregation, as mentioned above, and also participates in controlling its ATPase activity [43][94]. Zhai et al. [95] showed that His107, conserved in all mitochondrial and chloroplast HSP70-escort proteins, is especially required for ATPase stimulation. In humans, HEP1 binds the mortalin NBD directly [94], while in yeast, an interdomain linker is needed [43]. As noted above, the interdomain linker is a short loop of hydrophobic amino acids that connect the mtHSP70 NBD to its SBD, providing for their mutual communication [96]. Interestingly, the presence of human HEP1 increases the mtHSP70 ATPase activity by up to 49-fold [97], similar to the effect reported for the J-domain co-chaperones [98][44], and it also enhances the rate of nucleotide exchange, similar to the GRPE-type co-chaperones [47].
4.2 GRPE
In complex with mtHSP70, GRPE acts as a nucleotide exchange factor (NEF) (Figure 2), mediating the opening of the HSP70 nucleotide binding cleft to facilitate the dissociation of ADP, which, in turn, allows the binding of another ATP molecule and promoting the release of the protein substrate [99][100].
In E. coli , GrpE acts as a NEF for the bacterial Hsp70 homologue DnaK [47]. In S. cerevisiae , MGE1 acts as a NEF for the yeast mtHSP70 SSC1 [101]. Mammals have two mitochondrial GRPE homologues, GRPEL1 (a 23 kDa protein) and GRPEL2 (25 kDa) [102] together with an additional exchange factor, BAG1, for cytosolic HSP70 [103]. The actual reason for such duplicity in mitochondria is still unknown, though several experiments have shown that GRPEL1 is essential for cell viability while GRPEL2 is not [104][105][106]. Konovalova et al. [106] found that GRPEL1 serves as the major exchange factor for mtHSP70 (mortalin), which is necessary for maintaining the proper translocation of proteins to the mitochondrial matrix through the PAM complex, while GRPEL2 acts as a “helper” protein. Konovalova et al. [106] also found that human GRPEL2 is a redox-sensitive protein, which can form dimers (through disulfide linkages) under oxidative stress. This suggests that even though GRPEL2 is not essential in cultured cells, it has may have adapted to fine-tune protein import and folding in response to altered redox conditions.
4.3 TID-1
TID-1 (tumorous imaginal disc protein 1), also known as DnaJ homolog subfamily A member 3 (DnaJA3), is a small mitochondrial protein belonging to the HSP40 sub-family. TID-1 is a human homologue of the bacterial protein DnaJ and the tumor suppressor protein TID-56 present in Drosophila [107][108]. Its translation occurs in the cytosol, and mRNA splicing produces two isoforms: a 43 kDa long isoform called TID-1L, and a shorter 40 kDa isoform called TID-1S. These isoforms differ in the length of their C-terminal ends (33 residues for TID-1L and 6 for TID-1S) [109][110]. NMR studies have shown that TID-1 contains a highly conserved J-domain at its N-terminus, typical for all DnaJ proteins, which is extremely important for its binding to mtHSP70 [111][112][48]. TID‐1L resides mainly in the cytosol, where it interacts with cytosolic HSP70 and participates in the induction of apoptosis and various cellular signaling pathways. TID-1S, on the other hand, localizes primarily to the mitochondrial matrix, where it is responsible for mtDNA stability and maintains the mitochondrial membrane potential, thus acting against apoptosis [113][114][115][116]. TID-1 is one of the major co-chaperones of mtHSP70 and is mainly involved in stimulating its ATPase activity (Figure 2). Indeed, when other HSP70 co-chaperones are missing, a TID-1–mtHSP70 complex can still bind unfolded substrates and prevent their aggregation [117].
TID-1 is involved in numerous cellular processes, including cell growth, proliferation, differentiation, ageing, and survival [118][119][120][121]. In mammals, it is also involved in movement and plays an important role in the development of embryos and skeletal muscles [122][123][124]. Loss of TID-1 in heart results in cardiomyopathies and a decrease in mtDNA levels because it aids in the proper folding of DNA polymerase γ [125]. The protein is also involved in regulating mitochondrial homeostasis. Deregulation of TID-1 impairs its interaction with the mitochondrial dynamin-1-like protein DNM1L, causing fragmentation of the mitochondrial network [119] and negatively influences the CR6-interacting factor 1 (CRIF1) involved in the proper localization of the OXPHOS subunits into the inner mitochondrial membrane [126]. Similarly, an imbalance between TID-1 and mtHSP70 may be responsible for the mitochondrial fragmentation seen in patients diagnosed with optic atrophy 1 [127]. A recent study by Patra et al. [128] linked ataxia and developmental delay in one patient to a homozygous mutation in the TID1 gene (Arg151Thr), which produced a TID-1 variant that was less efficiently imported into the mitochondria and had severely impaired co-chaperone functions.
5. Conclusions
The mitochondrial HSP70 chaperone system consisting of mortalin and its co‐chaperones HEP1, TID‐1, and GRPE, forms one of the key components of PQC in mitochondria. As an essential protein that participates in proliferation, functional maintenance, and cellular stress response, mortalin has been implicated in many human pathologies, including neurodegenerative disorders, autosomal recessive diseases, and carcinogenesis. Indeed, recent studies indicate that mortalin could serve as a prognostic and predictive marker of cancer invasiveness as its up‐regulation has been detected in a variety of malignancies, such as brain tumors, hepatocellular carcinoma, colon carcinoma, breast cancer, and leukemia.
References
- Bertelsen, E.B.; Chang, C.Y.; Gestwicki, J.E.; Zuiderweg, E.R.P. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. USA 2009, 106, 8471–8476.
- Kityk, R.; Kopp, J.; Sinning, I.; Mayer, M. Structure and Dynamics of the ATP-Bound Open Conformation of Hsp70 Chaperones. Mol. Cell 2012, 48, 863–874.
- Fernández-Fernández, M.R.; Gragera, M.; Ochoa-Ibarrola, L.; Quintana-Gallardo, L.; Valpuesta, J. Hsp70—A master regulator in protein degradation. FEBS Lett. 2017, 591, 2648–2660.
- Dores-Silva, P.R.; Nishimura, L.S.; Kiraly, V.T.; Borges, J.C. Structural and functional studies of the Leishmania braziliensis mitochondrial Hsp70: Similarities and dissimilarities to human orthologues. Arch. Biochem. Biophys. 2017, 613, 43–52.
- Saibil, H.R. The PDB and protein homeostasis: From chaperones to degradation and disaggregase machines. J. Biol. Chem. 2021, 296, 100744.
- Voos, W. Chaperone–protease networks in mitochondrial protein homeostasis. Biochim. Biophys. Acta 2013, 1833, 388–399.
- Da Silva, K.P. The Molecular Chaperone Hsp70 Family Members Function by a Bidirectional Heterotrophic Allosteric Mechanism. Protein Pept. Lett. 2011, 18, 132–142.
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680.
- Velasco, L.; Dublang, L.; Moro, F.; Muga, A. The Complex Phosphorylation Patterns that Regulate the Activity of Hsp70 and Its Cochaperones. Int. J. Mol. Sci. 2019, 20, 4122.
- Zuiderweg, E.R.P.; Bertelsen, E.B.; Rousaki, A.; Mayer, M.; Gestwicki, J.E.; Ahmad, A. Allostery in the Hsp70 Chaperone Proteins. Top. Curr. Chem. 2012, 328, 99–153.
- Mayer, M.P. Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem. Sci. 2013, 38, 507–514.
- Dores-Silva, P.R.; Barbosa, L.; Ramos, C.; Borges, J.C. Human Mitochondrial Hsp70 (Mortalin): Shedding Light on ATPase Activity, Interaction with Adenosine Nucleotides, Solution Structure and Domain Organization. PLoS ONE 2015, 10, e0117170.
- Wadhwa, R.; Kaul, S.; Ikawa, Y.; Sugimoto, Y. Identification of a novel member of mouse hsp70 family. Its association with cellular mortal phenotype. J. Biol. Chem. 1993, 268, 6615–6621.
- Naylor, D.J.; Hoogenraad, N.J.; Høj, P.B. Isolation and characterisation of a cDNA encoding rat mitochondrial GrpE, a stress-inducible nucleotide-exchange factor of ubiquitous appearance in mammalian organs. FEBS Lett. 1996, 396, 181–188.
- Kaul, S.; Deocaris, C.C.; Wadhwa, R. Three faces of mortalin: A housekeeper, guardian and killer. Exp. Gerontol. 2007, 42, 263–274.
- Deocaris, C.C.; Kaul, S.C.; Wadhwa, R. From proliferative to neurological role of an hsp70 stress chaperone, mortalin. Biogerontology 2008, 9, 391–403.
- Deocaris, C.C.; Widodo, N.; Ishii, T.; Kaul, S.C.; Wadhwa, R. Functional Significance of Minor Structural and Expression Changes in Stress Chaperone Mortalin. Ann. N. Y. Acad. Sci. 2007, 1119, 165–175.
- Deocaris, C.C.; Widodo, N.; Shrestha, B.G.; Kaur, K.; Ohtaka, M.; Yamasaki, K.; Kaul, S.C.; Wadhwa, R. Mortalin sensitizes human cancer cells to MKT-077-induced senescence. Cancer Lett. 2007, 252, 259–269.
- Wadhwa, R.; Yaguchi, T.; Hasan, K.; Taira, K.; Kaul, S.C. Mortalin–MPD (mevalonate pyrophosphate decarboxylase) interactions and their role in control of cellular proliferation. Biochem. Biophys. Res. Commun. 2003, 302, 735–742.
- Takano, S.; Wadhwa, R.; Mitsui, Y.; Kaul, S.C. Identification and characterization of molecular interactions between glucose-regulated proteins (GRPs) mortalin/GRP75/peptide-binding protein 74 (PBP74) and GRP94. Biochem. J. 2001, 357, 393–398.
- Dores-Silva, P.; Minari, K.; Ramos, C.; Barbosa, L.; Borges, J. Structural and stability studies of the human mtHsp70-escort protein 1: An essential mortalin co-chaperone. Int. J. Biol. Macromol. 2013, 56, 140–148.
- Vu, M.T.; Zhai, P.; Lee, J.; Guerra, C.; Liu, S.; Gustin, M.C.; Silberg, J.J. The DNLZ/HEP zinc-binding subdomain is critical for regulation of the mitochondrial chaperone HSPA9. Protein Sci. 2012, 21, 258–267.
- Harrison, C.J.; Hayer-Hartl, M.; Di Liberto, M.; Hartl, F.-U.; Kuriyan, J. Crystal Structure of the Nucleotide Exchange Factor GrpE Bound to the ATPase Domain of the Molecular Chaperone DnaK. Science 1997, 276, 431–435.
- Harrison, C. GrpE, a nucleotide exchange factor for DnaK. Cell Stress Chaperon 2003, 8, 218–224.
- Syken, J.; De-Medina, T.; Münger, K. TID1, a human homolog of the Drosophila tumor suppressor l(2)tid, encodes two mitochondrial modulators of apoptosis with opposing functions. Proc. Natl. Acad. Sci. USA 1999, 96, 8499–8504.
- Liu, T.; Daniels, C.K.; Cao, S. Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol. Ther. 2012, 136, 354–374.
- Ng, A.C.-H.; Baird, S.D.; Screaton, R.A. Essential Role of TID1 in Maintaining Mitochondrial Membrane Potential Homogeneity and Mitochondrial DNA Integrity. Mol. Cell. Biol. 2014, 34, 1427–1437.
- Lu, B.; Garrido, N.; Spelbrink, J.N.; Suzuki, C.K. Tid1 Isoforms Are Mitochondrial DnaJ-like Chaperones with Unique Carboxyl Termini That Determine Cytosolic Fate. J. Biol. Chem. 2006, 281, 13150–13158.
- Iosefson, O.; Sharon, S.; Goloubinoff, P.; Azem, A. Reactivation of protein aggregates by mortalin and Tid1—The human mitochondrial Hsp70 chaperone system. Cell Stress Chaperon 2011, 17, 57–66.
- Yoshimune, K.; Yoshimura, T.; Esaki, N. Hsc62, a New DnaK Homologue ofEscherichia coli. Biochem. Biophys. Res. Commun. 1998, 250, 115–118.
- Kluck, C.J.; Patzelt, H.; Genevaux, P.; Brehmer, D.; Rist, W.; Schneider-Mergener, J.; Bukau, B.; Mayer, M. Structure-Function Analysis of HscC, theEscherichia coli Member of a Novel Subfamily of Specialized Hsp70 Chaperones. J. Biol. Chem. 2002, 277, 41060–41069.
- Kominek, J.; Marszalek, J.; Neuvéglise, C.; Craig, E.A.; Williams, B.L. The Complex Evolutionary Dynamics of Hsp70s: A Genomic and Functional Perspective. Genome Biol. Evol. 2013, 5, 2460–2477.
- Takakuwa, J.E.; Nitika; Knighton, L.E.; Truman, A.W. Oligomerization of Hsp70: Current Perspectives on Regulation and Function. Front. Mol. Biosci. 2019, 6, 81.
- Kabani, M.; Martineau, C.N. Multiple Hsp70 Isoforms in the Eukaryotic Cytosol: Mere Redundancy or Functional Specificity? Curr. Genom. 2008, 9, 338-248.
- Bonam, S.R.; Ruff, M.; Muller, S. Ruff HSPA8/HSC70 in Immune Disorders: A Molecular Rheostat that Adjusts Chaperone-Mediated Autophagy Substrates. Cells 2019, 8, 849.
- Velasco, L.; Dublang, L.; Moro, F.; Muga, A. The Complex Phosphorylation Patterns that Regulate the Activity of Hsp70 and Its Cochaperones. Int. J. Mol. Sci. 2019, 20, 4122.
- General, I.J.; Liu, Y.; Blackburn, M.E.; Mao, W.; Gierasch, L.M.; Bahar, I. ATPase Subdomain IA Is a Mediator of Interdomain Allostery in Hsp70 Molecular Chaperones. PLoS Comput. Biol. 2014, 10, e1003624.
- Kityk, R.; Vogel, M.; Schlecht, R.; Bukau, B.; Mayer, M.P. Pathways of allosteric regulation in Hsp70 chaperones. Nat. Commun. 2015, 6, 8308.
- Morshauser, R.C.; Wang, H.; Flynn, G.C.; Zuiderweg, E.R.P. The Peptide-Binding Domain of the Chaperone Protein Hsc70 Has an Unusual Secondary Structure Topology. Biochemistry 1995, 34, 6261–6266.
- Zhu, X.; Zhao, X.; Burkholder, W.F.; Gragerov, A.; Ogata, C.M.; Gottesman, M.E.; Hendrickson, W.A. Structural Analysis of Substrate Binding by the Molecular Chaperone DnaK. Science 1996, 272, 1606–1614.
- Da Silva, K.P. The Molecular Chaperone Hsp70 Family Members Function by a Bidirectional Heterotrophic Allosteric Mechanism. Protein Pept. Lett. 2011, 18, 132–142.
- Zuiderweg, E.R.P.; Bertelsen, E.B.; Rousaki, A.; Mayer, M.; Gestwicki, J.E.; Ahmad, A. Allostery in the Hsp70 Chaperone Proteins. Top. Curr. Chem. 2012, 328, 99–153.
- Mayer, M.P. Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem. Sci. 2013, 38, 507–514.
- Dores-Silva, P.R.; Barbosa, L.; Ramos, C.; Borges, J.C. Human Mitochondrial Hsp70 (Mortalin): Shedding Light on ATPase Activity, Interaction with Adenosine Nucleotides, Solution Structure and Domain Organization. PLoS ONE 2015, 10, e0117170.
- Kaul, S.C.; Wadhwa, R. Mortalin Biology: Life, Stress and Death; Springer: Dordrecht, The Netherlands, 2012; p. 342.
- Amick, J.; Schlanger, S.E.; Wachnowsky, C.; Moseng, M.A.; Emerson, C.C.; Dare, M.; Luo, W.-I.; Ithychanda, S.S.; Nix, J.C.; Cowan, J.A.; et al. Crystal structure of the nucleotide-binding domain of mortalin, the mitochondrial Hsp70 chaperone. Protein Sci. 2014, 23, 833–842.
- Giometti, C.S.; Tollaksen, S.L.; Chubb, C.; Williams, C.; Huberman, E. Analysis of proteins from human breast epithelial cells using two-dimensional gel electrophoresis. Electrophoresis 1995, 16, 1215–1224.
- Hochstrasser, D.F.; Frutiger, S.; Paquet, N.; Bairoch, A.; Ravier, F.; Pasquali, C.; Sanchez, J.-C.; Tissot, J.-D.; Bjellqvist, B.; Vargas, R.; et al. Human liver protein map: A reference database established by microsequencing and gel comparison. Electrophoresis 1992, 13, 992–1001.
- Ji, H.; Reid, G.E.; Moritz, R.L.; Eddes, J.S.; Burgess, A.W.; Simpson, R.J. A two-dimensional gel database of human colon carcinoma proteins. Electrophoresis 1997, 18, 605–613.
- Sun, J.; Che, S.-L.; Piao, J.-J.; Xu, M.; Chen, L.-Y.; Lin, Z.-H. Mortalin overexpression predicts poor prognosis in early stage of non–small cell lung cancer. Tumor Biol. 2017, 39.
- Bohnert, M.; Pfanner, N.; van der Laan, M. A dynamic machinery for import of mitochondrial precursor proteins. FEBS Lett. 2007, 581, 2802–2810.
- Dolezal, P.; Likic, V.; Tachezy, J.; Lithgow, T. Evolution of the Molecular Machines for Protein Import into Mitochondria. Science 2006, 313, 314–318.
- Mokranjac, D.; Neupert, W. Thirty years of protein translocation into mitochondria: Unexpectedly complex and still puzzling. Biochim. Biophys. Acta 2009, 1793, 33–41.
- Kang, P.-J.; Ostermann, J.; Shilling, J.; Neupert, W.; Craig, E.A.; Pfanner, N. Requirement for hsp70 in the mitochondrial matrix for translocation and folding of precursor proteins. Nat. Cell Biol. 1990, 348, 137–143.
- Rehling, P.; Brandner, K.; Pfanner, N. Mitochondrial import and the twin-pore translocase. Nat. Rev. Mol. Cell Biol. 2004, 5, 519–530.
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284.
- Van Wilpe, S.; Ryan, M.; Hill, K.; Maarse, A.C.; Meisinger, C.; Brix, J.; Dekker, P.; Moczko, M.; Wagner, R.; Meijer, M.; et al. Tom22 is a multifunctional organizer of the mitochondrial preprotein translocase. Nat. Cell Biol. 1999, 401, 485–489.
- Abe, Y.; Shodai, T.; Muto, T.; Mihara, K.; Torii, H.; Nishikawa, S.-I.; Endo, T.; Kohda, D. Structural Basis of Presequence Recognition by the Mitochondrial Protein Import Receptor Tom20. Cell 2000, 100, 551–560.
- Dekker, P.J.; Keil, P.; Rassow, J.; Maarse, A.C.; Pfanner, N.; Meijer, M. Identification of MIM23, a putative component of the protein import machinery of the mitochondrial inner membrane. FEBS Lett. 1993, 330, 66–70.
- Lohret, T.A.; Jensen, R.E.; Kinnally, K.W. Tim23, a Protein Import Component of the Mitochondrial Inner Membrane, Is Required for Normal Activity of the Multiple Conductance Channel, MCC. J. Cell Biol. 1997, 137, 377–386.
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714.
- Dores-Silva, P.R.; Cauvi, D.M.; Kiraly, V.T.; Borges, J.C.; De Maio, A. Human HSPA9 (mtHsp70, mortalin) interacts with lipid bilayers containing cardiolipin, a major component of the inner mitochondrial membrane. Biochim. Biophys. Acta 2020, 1862, 183436.
- Bukau, B.; Horwich, A.L. The Hsp70 and Hsp60 Chaperone Machines. Cell 1998, 92, 351–366.
- Dutkiewicz, R.; Schilke, B.; Knieszner, H.; Walter, W.; Craig, E.A.; Marszalek, J.; Oinuma, K.-I.; Hashimoto, Y.; Konishi, K.; Goda, M.; et al. Ssq1, a Mitochondrial Hsp70 Involved in Iron-Sulfur (Fe/S) Center Biogenesis. J. Biol. Chem. 2003, 278, 29719–29727.
- Schilke, B.; Williams, B.; Knieszner, H.; Pukszta, S.; D’Silva, P.; Craig, E.A.; Marszalek, J. Evolution of Mitochondrial Chaperones Utilized in Fe-S Cluster Biogenesis. Curr. Biol. 2006, 16, 1660–1665.
- Shan, Y.; Cortopassi, G. Mitochondrial Hspa9/Mortalin regulates erythroid differentiation via iron-sulfur cluster assembly. Mitochondrion 2016, 26, 94–103.
- Altieri, D.C.; Stein, G.S.; Lian, J.B.; Languino, L. TRAP-1, the mitochondrial Hsp90. Biochim. Biophys. Acta 2012, 1823, 767–773.
- Felts, S.J.; Owen, B.A.L.; Nguyen, P.; Trepel, J.; Donner, D.B.; Toft, D.O. The hsp90-related Protein TRAP1 Is a Mitochondrial Protein with Distinct Functional Properties. J. Biol. Chem. 2000, 275, 3305–3312.
- Schwarzer, C.; Barnikol-Watanabe, S.; Thinnes, F.P.; Hilschmann, N. Voltage-dependent anion-selective channel (VDAC) interacts with the dynein light chain Tctex1 and the heat-shock protein PBP74. Int. J. Biochem. Cell Biol. 2002, 34, 1059–1070.
- Liu, Y.; Liu, W.; Song, X.-D.; Zuo, J. Effect of GRP75/mthsp70/PBP74/mortalin overexpression on intracellular ATP level, mitochondrial membrane potential and ROS accumulation following glucose deprivation in PC12 cells. Mol. Cell. Biochem. 2005, 268, 45–51.
- Burbulla, L.F.; Schelling, C.; Kato, H.; Rapaport, D.; Woitalla, D.; Schiesling, C.; Schulte, C.; Sharma, M.; Illig, T.; Bauer, P.; et al. Dissecting the role of the mitochondrial chaperone mortalin in Parkinson’s disease: Functional impact of disease-related variants on mitochondrial homeostasis. Hum. Mol. Genet. 2010, 19, 4437–4452.
- Alkhaja, A.K.; Jans, D.C.; Nikolov, M.; Vukotic, M.; Lytovchenko, O.; Ludewig, F.; Schliebs, W.; Riedel, D.; Urlaub, H.; Jakobs, S.; et al. MINOS1 is a conserved component of mitofilin complexes and required for mitochondrial function and cristae organization. Mol. Biol. Cell 2012, 23, 247–257.
- Bogenhagen, D.F.; Rousseau, D.; Burke, S. The Layered Structure of Human Mitochondrial DNA Nucleoids. J. Biol. Chem. 2008, 283, 3665–3675.
- Sichting, M.; Mokranjac, D.; Azem, A.; Neupert, W.; Hell, K. Maintenance of structure and function of mitochondrial Hsp70 chaperones requires the chaperone Hep1. EMBO J. 2005, 24, 1046–1056.
- Goswami, A.V.; Chittoor, B.; D’Silva, P. Understanding the Functional Interplay between Mammalian Mitochondrial Hsp70 Chaperone Machine Components. J. Biol. Chem. 2010, 285, 19472–19482.
- Zhai, P.; Vu, M.T.; Hoff, K.G.; Silberg, J.J. A conserved histidine in human DNLZ/HEP is required for stimulation of HSPA9 ATPase activity. Biochem. Biophys. Res. Commun. 2011, 408, 589–594.
- Blamowska, M.; Sichting, M.; Mapa, K.; Mokranjac, D.; Neupert, W.; Hell, K. ATPase Domain and Interdomain Linker Play a Key Role in Aggregation of Mitochondrial Hsp70 Chaperone Ssc1. J. Biol. Chem. 2010, 285, 4423–4431.
- Zhai, P.; Stanworth, C.; Liu, S.; Silberg, J.J. The Human Escort Protein Hep Binds to the ATPase Domain of Mitochondrial Hsp70 and Regulates ATP Hydrolysis. J. Biol. Chem. 2008, 283, 26098–26106.
- Silberg, J.J.; Tapley, T.L.; Hoff, K.G.; Vickery, L.E. Regulation of the HscA ATPase Reaction Cycle by the Co-chaperone HscB and the Iron-Sulfur Cluster Assembly Protein IscU. J. Biol. Chem. 2004, 279, 53924–53931.
- Misselwitz, B.; Staeck, O.; A Rapoport, T. J Proteins Catalytically Activate Hsp70 Molecules to Trap a Wide Range of Peptide Sequences. Mol. Cell 1998, 2, 593–603.
- Liberek, K.; Marszalek, J.; Ang, D.; Georgopoulos, C.; Zylicz, M. Escherichia coli DnaJ and GrpE heat shock proteins jointly stimulate ATPase activity of DnaK. Proc. Natl. Acad. Sci. USA 1991, 88, 2874–2878.
- Moro, F.; Muga, A. Thermal Adaptation of the Yeast Mitochondrial Hsp70 System is Regulated by the Reversible Unfolding of its Nucleotide Exchange Factor. J. Mol. Biol. 2006, 358, 1367–1377.
- Naylor, D.J.; Stines, A.P.; Hoogenraad, N.J.; Høj, P.B. Evidence for the Existence of Distinct Mammalian Cytosolic, Microsomal, and Two Mitochondrial GrpE-like Proteins, the Co-chaperones of Specific Hsp70 Members. J. Biol. Chem. 1998, 273, 21169–21177.
- Takayama, S.; Bimston, D.N.; Matsuzawa, S.; Freeman, B.C.; Aimé-Sempé, C.; Xie, Z.; Morimoto, R.I.; Reed, J.C. BAG-1 modulates the chaperone activity of Hsp70/Hsc70. EMBO J. 1997, 16, 4887–4896.
- Ikeda, E.; Yoshida, S.; Mitsuzawa, H.; Uno, I.; Toh-E, A. YGE1is a yeast homologue ofEscherichia coli grpEand is required for maintenance of mitochondrial functions. FEBS Lett. 1994, 339, 265–268.
- Srivastava, S.; Savanur, M.A.; Sinha, D.; Birje, A.; Vigneshwaran, R.; Saha, P.P.; D’Silva, P. Regulation of mitochondrial protein import by the nucleotide exchange factors GrpEL1 and GrpEL2 in human cells. J. Biol. Chem. 2017, 292, 18075–18090.
- Konovalova, S.; Liu, X.; Manjunath, P.; Baral, S.; Neupane, N.; Hilander, T.; Yang, Y.; Balboa, D.; Terzioglu, M.; Euro, L.; et al. Redox regulation of GRPEL2 nucleotide exchange factor for mitochondrial HSP70 chaperone. Redox Biol. 2018, 19, 37–45.
- Schilling, B.; De-Medina, T.; Syken, J.; Vidal, M.; Munger, K. A Novel Human DnaJ Protein, hTid-1, a Homolog of the Drosophila Tumor Suppressor Protein Tid56, Can Interact with the Human Papillomavirus Type 16 E7 Oncoprotein. Virology 1998, 247, 74–85.
- Wang, T.-H.; Lin, Y.-H.; Yang, S.-C.; Chang, P.-C.; Wang, T.-C.; Chen, C.-Y. Tid1-S regulates the mitochondrial localization of EGFR in non-small cell lung carcinoma. Oncogenesis 2017, 6, e361.
- Iosefson, O.; Sharon, S.; Goloubinoff, P.; Azem, A. Reactivation of protein aggregates by mortalin and Tid1—The human mitochondrial Hsp70 chaperone system. Cell Stress Chaperon 2011, 17, 57–66.
- Syken, J.; De-Medina, T.; Münger, K. TID1, a human homolog of the Drosophila tumor suppressor l(2)tid, encodes two mitochondrial modulators of apoptosis with opposing functions. Proc. Natl. Acad. Sci. USA 1999, 96, 8499–8504.
- Cyr, D.M.; Langer, T.; Douglas, M.G. DnaJ-like proteins: Molecular chaperones and specific regulators of Hsp70. Trends Biochem. Sci. 1994, 19, 176–181.
- Georgopoulos, C.; Welch, W.J. Role of the Major Heat Shock Proteins as Molecular Chaperones. Annu. Rev. Cell Biol. 1993, 9, 601–634.
- Silver, P.A.; Way, J.C. Eukaryotic DnaJ homologs and the specificity of Hsp70 activity. Cell 1993, 74, 5–6.
- Tarunina, M.; Alger, L.; Chu, G.; Munger, K.; Gudkov, A.; Jat, P.S. Functional Genetic Screen for Genes Involved in Senescence: Role of Tid1, a Homologue of the Drosophila Tumor Suppressor l(2)tid, in Senescence and Cell Survival. Mol. Cell. Biol. 2004, 24, 10792–10801.
- Elwi, A.N.; Lee, B.; Meijndert, H.C.; Braun, J.E.; Kim, S.-W. Mitochondrial chaperone DnaJA3 induces Drp1-dependent mitochondrial fragmentation. Int. J. Biochem. Cell Biol. 2012, 44, 1366–1376.
- Trinh, D.L.; Elwi, A.N.; Kim, S.-W. Direct interaction between p53 and Tid1 proteins affects p53 mitochondrial localization and apoptosis. Oncotarget 2010, 1, 396–404.
- Ahn, B.Y.; Trinh, D.L.N.; Zajchowski, L.D.; Lee, B.; Elwi, A.N.; Kim, S.-W. Tid1 is a new regulator of p53 mitochondrial translocation and apoptosis in cancer. Oncogene 2009, 29, 1155–1166.
- Lo, J.-F.; Hayashi, M.; Woo-Kim, S.; Tian, B.; Huang, J.-F.; Fearns, C.; Takayama, S.; Zapata, J.M.; Yang, Y.; Lee, J.-D. Tid1, a Cochaperone of the Heat Shock 70 Protein and the Mammalian Counterpart of the Drosophila Tumor Suppressor l(2)tid, Is Critical for Early Embryonic Development and Cell Survival. Mol. Cell. Biol. 2004, 24, 2226–2236.
- Choi, J.H.; Choi, D.-K.; Sohn, K.-C.; Kwak, S.S.; Suk, J.; Lim, J.-S.; Shin, I.; Kim, S.-W.; Lee, J.-H.; Joe, C.O. Absence of a Human DnaJ Protein hTid-1S Correlates with Aberrant Actin Cytoskeleton Organization in Lesional Psoriatic Skin. J. Biol. Chem. 2012, 287, 25954–25963.
- Cheng, L.-H.; Hung, K.-F.; Lee, T.-C.; Huang, C.-Y.; Chiu, W.-T.; Lo, J.-F.; Huang, T.-F. Mitochondrial co-chaperone protein Tid1 is required for energy homeostasis during skeletal myogenesis. Stem Cell Res. Ther. 2016, 7, 185.
- Hayashi, M.; Imanaka-Yoshida, K.; Yoshida, T.; Wood, M.; Fearns, C.; Tatake, R.J.; Lee, J.-D. A crucial role of mitochondrial Hsp40 in preventing dilated cardiomyopathy. Nat. Med. 2005, 12, 128–132.
- Kim, S.J.; Kwon, M.-C.; Ryu, M.J.; Chung, H.K.; Tadi, S.; Kim, Y.K.; Kim, J.M.; Lee, S.H.; Park, J.H.; Kweon, G.R.; et al. CRIF1 Is Essential for the Synthesis and Insertion of Oxidative Phosphorylation Polypeptides in the Mammalian Mitochondrial Membrane. Cell Metab. 2012, 16, 274–283.
- Lee, B.; Ahn, Y.; Kang, S.-M.; Park, Y.; Jeon, Y.-J.; Rho, J.M.; Kim, S.-W. Stoichiometric expression of mtHsp40 and mtHsp70 modulates mitochondrial morphology and cristae structure via Opa1L cleavage. Mol. Biol. Cell 2015, 26, 2156–2167.
- Patra, M.; Weiss, C.; Abu-Libdeh, B.; Ashhab, M.; Abuzer, S.; Elpeleg, O.; Mahajnah, M.; Kessel, A.; Azem, A. A novel variant of the human mitochondrial DnaJ protein, Tid1, associates with a human disease exhibiting developmental delay and polyneuropathy. Eur. J. Hum. Genet. 2019, 27, 1072–1080.

