 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dmitry Borisovich Zorov | + 2282 word(s) | 2282 | 2021-08-09 11:19:19 | | | |
| 2 | Ron Wang | -108 word(s) | 2174 | 2021-08-26 06:07:19 | | | | |
| 3 | Ron Wang | + 76 word(s) | 2250 | 2021-08-26 06:08:05 | | |
Video Upload Options
The term uncoupling is used concerning the situation when a proton bypasses ATP synthase and electron transport becomes disconnected from the process of ATP synthesis due to a short-circuit of the membrane potential existing across the membrane. It also applies to mitochondria, where uncoupling causes a loss of the tight association of oxidation of respiratory substrates and ATP synthesis. Generally speaking, uncouplers exert their action by organizing a proton leak in the inner mitochondrial membrane, which jeopardizes the generation of the proton motive force.
1. Introduction
There is a great cluster of knowledge about the protective capabilities of uncoupling of oxidative phosphorylation in the mitochondria of numerous cells, including neural ones, thus compromising chemiosmotic mechanism of energy production [1][2][3][4][5][6][7][8][9][10][11]. According to this mechanism, proton pumps residing in coupling membranes generate the transmembrane potential of hydrogen ions (protons), which is used by ATP synthase to make ATP, thus organizing a tight coupling between the processes of oxidation and phosphorylation. The requisite of this mechanism is a tightly regulated proton cycling existing across the coupling membrane and resulting in a high yield of ATP. The term uncoupling is used concerning the situation when a proton bypasses ATP synthase and electron transport becomes disconnected from the process of ATP synthesis due to a short-circuit of the membrane potential existing across the membrane. It also applies to mitochondria, where uncoupling causes a loss of the tight association of oxidation of respiratory substrates and ATP synthesis [12][13]. Generally speaking, uncouplers exert their action by organizing a proton leak in the inner mitochondrial membrane, which jeopardizes the generation of the proton motive force [14]. Limited usage of uncouplers was announced as one of the most efficient strategies to coupe with different pathologies including aging [15]. In subcellular, cellular and organismal experiments, three uncouplers are the most widely used: DNP (2,4-dinitrophenol; active concentrations are in the region of 100 µM), CCCP (carbonylcyanide-3-chlorophenylhydrazone; active concentrations are about two orders less than of DNP), and FCCP (carbonylcyanide-4-trifluoromethoxyphenylhydrazone; active concentrations are at least one order less than of CCCP).
At the current level of our knowledge on the mechanisms of uncoupling, two modes are distinguished. According to the first mechanism, even uncouplers using a bilayer membrane provide a proton leak, causing a collapse of the proton gradient through the membrane [13][16]. Data in recent years indicate the existence of another mechanism involving special proteins in the coupling membranes that mediate the action of uncouplers [17][18][19][20][21]), while the number of nominal candidate proteins for the role is constantly increasing. It should be noted that there is a fundamental difference between these proteins and natural uncoupling proteins (UCPs) [22] , the functioning of which differ from those mentioned.
Meanwhile, the impression arises that moderate uncoupling unambiguously contributes to better resistance to various internal and external challenges, which may attribute this method of manipulating mitochondrial activity to a neuroprotective strategy. However, almost nowhere are the negative aspects considered, which should always be evaluated when using uncouplers in experimental and clinical practice, especially for brain pathologies. Previously, we have already given a similar assessment, weighing all the pros and cons of using mitochondria-targeted antioxidants [23], which we will do in this mini-review discussing beneficial and disadvantageous factors accompanying the application of uncouplers.
2. Pros
The oxygen molecule, even in its triplet state, is a quite strong oxidizer, which, when in the cell, can provide unnecessary oxidation of essential molecules, such as proteins, lipids, and nucleic acids. There is a point of view that the very appearance on Earth of oxygen-utilizing bacteria, and later mitochondria, was evolutionarily dictated by the oxygen menace [24] due to the rise in oxygen in the atmosphere, which was called the Great Oxidation Event or the Great Oxygenation Event [25]. Normally, the supply of oxygen to the cell does not limit the rate of its utilization [26], and the measured values of pO 2 in the brain do not limit the activity of cytochrome oxidase, which is the main consumer of oxygen in tissue. By definition, uncouplers activate mitochondrial respiration, thereby potentially they reduce the intracellular values of pO 2. In case it does not reach critical levels when oxygen concentration does not limit respiration, such lowering of intracellular oxygen and thus its toxicity may be considered beneficial.
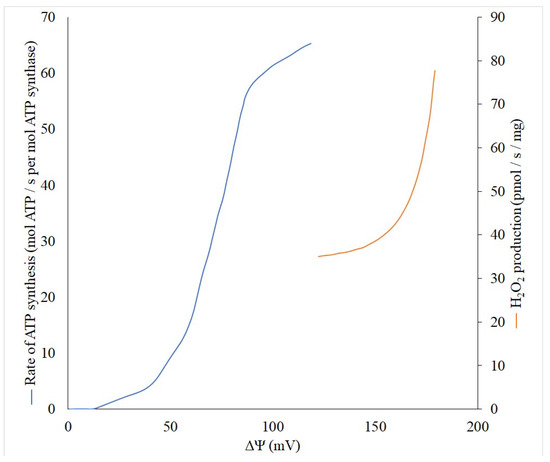
Theoretically, the production of reactive oxygen species (ROS) is a first-order reaction for oxygen, but there are some indicative exceptions, especially in the range of low pO2 values in tissue when an increased generation of ROS is observed [27][28]. However, the generation of ROS non-linearly depends on the membrane potential (Δψ) built in the inner mitochondrial membrane, while reaching an exponential character at high values of Δψ [29][30]. Thus, even a small decrease in the Δψ can lead to a significant reduction in the generation of ROS, and thereby reduce the risk of unnecessary oxidation of important cellular components. This behavior of ROS generation by mitochondria allowed the development of a strategy for combating pathologies accompanied by oxidative stress through using mild uncouplers. These compounds uncouple the processes of mitochondrial electron transport and phosphorylation, but in a very modest fashion, only slightly reducing the membrane potential, ultimately maintaining the ATP at a level that adequately meets metabolic demands. To understand the quantitative relationship between the membrane potential of mitochondria and their ability to generate ATP synthase and produce reactive oxygen species, we performed a literature search and linked these three parameters on one single graph ( Figure 1 ). Although it is impossible to fully match the data available in the literature, first, in both cases, there is a sigmoid dependence of the formation of ATP depending on the membrane potential with the presence of some threshold values, after which, a significant increase in generation begins. Secondly, even in the absence of experimental data for the full range of membrane potentials, it is clearly visible that after 120 mV the ATP generation reaches a plateau. This means that it is possible to reduce the membrane potential without significant violation of the energy balance up to 120–130 mV. Obviously, this can be used to define the concept of “mild” uncouplers, limiting their effective action to the ability to reduce the membrane potential but keeping it not lower than 120 mV. In accordance with modern knowledge, in general, the uncoupling effect of uncouplers which are lipophilic weak acids (p K a ∼4–8), is determined by their ability to be protonated on the side of the membrane where the proton concentration is higher, and translocated to another side. There, after dissociation, the bound proton is released, and the uncouplers return to their original location in the anionic form. The limiting stage of this entire cycle is the transmembrane transport of this anionic form [16]. It is important for uncoupling, which occurs with the participation of fatty acids, for example, caused by derivatives of Skulachev ions having the properties of mild uncouplers [31][32][33]. Thus, the strength (activity) of the uncouplers is partially determined by the rate of transport of the anionic form [16] and uncouplers potentially can be softly divided into two groups: strong and mild (weak), quantitatively discriminated by the active concentrations, from nanomolar to millimolar.
The activation of respiration caused by the use of uncouplers leads to increased mobilization of oxidative substrates and significant activation of oxidative metabolism, while, as the depletion of carbohydrate substrates occurs, the mobilization of fat resources takes place, which is desirable to combat obesity [34][35][36][37]. It should be noted that obesity is one of the risk factors for stroke, including in young adults [31]. Thus, therapeutic uncoupling can have an indirect neuroprotective effect through normalization of metabolism and improvement of the functioning of the cardiovascular system [38].
Uncouplers were found to significantly activate mitochondrial proliferation (biogenesis) [39][40], which is beneficial due to onset of compensatory mechanism designed to preserve mitochondrial ATP production under conditions of toxic mitochondrial damage. In experimental practice, mitochondrial biogenesis is most often associated with the activity of PGC-1α (transcriptional coactivator peroxisome proliferator activated receptor γ coactivator 1α [41], and usually, the proliferative mitochondrial activity is judged by the level of PGC-1α in the cell, which increases after the action of uncouplers. However, in adipocytes, mild mitochondrial uncoupling with FCCP did not stimulate mitochondrial biogenesis [42] which, firstly, raises the question of the lack of universality of the above-mentioned association and, secondly, may discriminate the process of powerful and moderate (mild) uncoupling, which is a function of a dose and chemical nature of an uncoupler. Moreover, while the uncoupler stimulated mitochondrial biogenesis in the oocytes of young animals, this did not happen in old animals [43], which points to another limiting factor that should be taken into account when evaluating the association of uncoupling and mitochondrial proliferation.
3. Cons
Uncoupler-induced lowering of the mitochondrial membrane potential retards all reactions driven by the membrane potential, of which there are many [44]. One of the most discussed functions (which is attributed to almost the main function that requires membrane potential homeostasis) is the directed transport of proteins into the mitochondria, without which the existence of the mitochondria itself is impossible, given that the mitochondrial genome provides only a small part of its protein needs [45][46][47][48].
The function of ensuring mitochondrial quality control is directly related to this mechanism, which includes the step of transport into the mitochondria of elements that control the quality of mitochondria [49] and the degradation of poorly functioning or non-functioning mitochondria to preserve a young and healthy phenotype [50][51].
A separate function that depends on the values of the membrane potential is the transport of ions into the mitochondria. Thermodynamically, the direction of the membrane potential ensures the transport of cations into the matrix and the exit of anions from it. Special importance is attached to the electrogenic transport of calcium ions in the mitochondria, the physiological significance of which is very high [52][53][54][55]. However, there are data that Ca 2+ overload and subsequent cell deterioration may be ameliorated by the use of uncouplers, which reduce the membrane potential-driven inward Ca 2+ transport in mitochondria [56][57]. Indeed, lowering mitochondrial membrane potential by uncoupling agents yielded a higher level of cell tolerance to cytosolic Ca 2+ overload caused by the neurotoxic effect of glutamate [58][59].
Although, as we have said, a large number of scientific teams are involved in studies of mitophagy over the world, some purely energetic elements of the process remain logically and actually unsupported and underexplored, and this primarily concerns the role of the mitochondrial membrane potential. The general statement about the mandatory presence of the mitochondrial membrane potential still stays. However, it is only qualitative in nature, while quantitative estimates of the necessary values of the membrane potential for mitophagy are practically absent. This differs the mitophagy process from ATP or peroxide generation presented in Figure 1 , where the threshold values, at which the processes practically do not occur, are obvious.
4. Current Insights
Our analysis, which can converge these two concepts, boils down to the fact that the second proposed mechanism is one of the examples of mitohormesis [60][61], stimulated by uncouplers. In this regard, a two-phase protective echelon can be implemented, which in the first stage, in the acute phase, reduces the high steady-state levels of ROS observed in the mentioned neurodegenerative states, but which can activate metabolism, with the possible short-term onset of near-ischemic states. These are accompanied by a short-term non-lethal increase in the level of ROS, triggering protective neuroprotective cascades, similar to those that occur during ischemic preconditioning [62][63][64][65].
The search for endogenous uncouplers has, so far, ended with the recognition that fatty acids and thyroid hormones can perform this function. This does not exclude mediators of the uncoupling process caused by uncoupling proteins (UCP 1–4, [22]), the full set of functions of which must be clarified, with the exception of UCP1, of which the main thermogenic function cannot be doubted. However, on the one hand, fatty acids can exhibit both direct uncoupling properties [66] or be mediators of transmembrane proteins: UCPs [67], translocator of adenine nucleotides (ANT) [68] and dicarboxylate carrier [18][69]. Other candidates for the role of an uncoupler, thyroxine and triiodothyronine (T3), have not been sufficiently investigated although in the last century there were indications of their uncoupling ability [70][71][72]. Among recent studies, work has demonstrated that T3 activates mitochondrial respiration via increased oxidation of fatty acids, while simultaneously enhancing autophagic flow and, in particular, mitophagy, deserves special attention [73]. The regulation of mitophagy by natural uncouplers (fatty acids and thyroxine) was also confirmed in experiments on cold exposure of animals, as a result of which the activation of mitophagy was observed in brown adipose tissue [74]. Later it was demonstrated that T3 stimulates brown adipose tissue through enhanced mitochondrial biogenesis and MTOR-mediated mitophagy [73].
Considering all these presented arguments, the extremely high metabolic activity of the brain requires a very careful approach for therapeutically induced manipulations, accompanied by an increase in the metabolism of neural cells, including neurons, astroglia, and endothelium. In general, the prevention of hypermetabolism of the brain is one of the immutable tasks that follows from knowledge of neuropathophysiology [75][76][77][78][79][80]. In general terms, this means that the therapeutic window of influence on brain metabolism is quite narrow, and if it is applied to uncouplers that increase metabolic activity, the therapeutic window of concentrations is also either very narrow or, due to its chemical nature, their uncoupling activity should be relatively small, accompanied by insignificant toxic properties.
References
- Fernanda, M.C.; Caldeira da Silva, C.C.; Fernanda, M.C.; Alicia, J.K. Mild Mitochondrial Uncoupling as a Therapeutic Strategy. Curr. Drug Targets 2011, 12, 783–789.
- Liu, D.; Zhang, Y.; Gharavi, R.; Park, H.R.; Lee, J.; Siddiqui, S.; Telljohann, R.; Nassar, M.R.; Cutler, R.G.; Becker, K.G.; et al. The Mitochondrial Uncoupler DNP Triggers Brain Cell MTOR Signaling Network Reprogramming and CREB Pathway Up-Regulation. J. Neurochem. 2015, 134, 677–692.
- Dorighello, G.G.; Rovani, J.C.; Paim, B.A.; Rentz, T.; Assis, L.H.P.; Vercesi, A.E.; Oliveira, H.C.F. Mild Mitochondrial Uncoupling Decreases Experimental Atherosclerosis, A Proof of Concept. J. Atheroscler. Thromb. 2021.
- Rai, Y.; Anita; Kumari, N.; Singh, S.; Kalra, N.; Soni, R.; Bhatt, A.N. Mild Mitochondrial Uncoupling Protects from Ionizing Radiation Induced Cell Death by Attenuating Oxidative Stress and Mitochondrial Damage. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148325.
- Berkowitz, B.A.; Olds, H.K.; Richards, C.; Joy, J.; Rosales, T.; Podolsky, R.H.; Childers, K.L.; Hubbard, W.B.; Sullivan, P.G.; Gao, S.; et al. Novel Imaging Biomarkers for Mapping the Impact of Mild Mitochondrial Uncoupling in the Outer Retina in Vivo. PLoS ONE 2020, 15, e0226840.
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795.
- Childress, E.S.; Alexopoulos, S.J.; Hoehn, K.L.; Santos, W.L. Small Molecule Mitochondrial Uncouplers and Their Therapeutic Potential. J. Med. Chem. 2018, 61, 4641–4655.
- Plotnikov, E.Y.; Silachev, D.N.; Jankauskas, S.S.; Rokitskaya, T.I.; Chupyrkina, A.A.; Pevzner, I.B.; Zorova, L.D.; Isaev, N.K.; Antonenko, Y.N.; Skulachev, V.P.; et al. Mild Uncoupling of Respiration and Phosphorylation as a Mechanism Providing Nephro- and Neuroprotective Effects of Penetrating Cations of the SkQ Family. Biochem. Mosc. 2012, 77, 1029–1037.
- Antonenko, Y.N.; Denisov, S.S.; Silachev, D.N.; Khailova, L.S.; Jankauskas, S.S.; Rokitskaya, T.I.; Danilina, T.I.; Kotova, E.A.; Korshunova, G.A.; Plotnikov, E.Y.; et al. A Long-Linker Conjugate of Fluorescein and Triphenylphosphonium as Mitochondria-Targeted Uncoupler and Fluorescent Neuro- and Nephroprotector. Biochim. Biophys. Acta 2016, 1860, 2463–2473.
- Silachev, D.N.; Khailova, L.S.; Babenko, V.A.; Gulyaev, M.V.; Kovalchuk, S.I.; Zorova, L.D.; Plotnikov, E.Y.; Antonenko, Y.N.; Zorov, D.B. Neuroprotective Effect of Glutamate-Substituted Analog of Gramicidin a Is Mediated by the Uncoupling of Mitochondria. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 3434–3442.
- Ost, M.; Keipert, S.; Klaus, S. Targeted Mitochondrial Uncoupling beyond UCP1—The Fine Line between Death and Metabolic Health. Biochimie 2017, 134, 77–85.
- Mitchell, P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic Type of Mechanism. Nature 1961, 191, 144–148.
- Liberman, E.A.; Topaly, V.P.; Tsofina, L.M.; Jasaitis, A.A.; Skulachev, V.P. Mechanism of Coupling of Oxidative Phosphorylation and the Membrane Potential of Mitochondria. Nature 1969, 222, 1076–1078.
- Skulachev, V.P. Uncoupling: New Approaches to an Old Problem of Bioenergetics. Biochim. Biophys. Acta 1998, 1363, 100–124.
- Caldeira da Silva, C.C.; Cerqueira, F.M.; Barbosa, L.F.; Medeiros, M.H.G.; Kowaltowski, A.J. Mild Mitochondrial Uncoupling in Mice Affects Energy Metabolism, Redox Balance and Longevity. Aging Cell 2008, 7, 552–560.
- McLaughlin, S.G.; Dilger, J.P. Transport of Protons across Membranes by Weak Acids. Physiol. Rev. 1980, 60, 825–863.
- Andreyev, A.Y.; Bondareva, T.O.; Dedukhova, V.I.; Mokhova, E.N.; Skulachev, V.P.; Volkov, N.I. Carboxyatractylate Inhibits the Uncoupling Effect of Free Fatty Acids. FEBS Lett. 1988, 226, 265–269.
- Samartsev, V.N.; Smirnov, A.V.; Zeldi, I.P.; Markova, O.V.; Mokhova, E.N.; Skulachev, V.P. Involvement of Aspartate/Glutamate Antiporter in Fatty Acid-Induced Uncoupling of Liver Mitochondria. Biochim. Biophys. Acta (BBA) Bioenerg. 1997, 1319, 251–257.
- Firsov, A.M.; Popova, L.B.; Khailova, L.S.; Nazarov, P.A.; Kotova, E.A.; Antonenko, Y.N. Protonophoric Action of BAM15 on Planar Bilayers, Liposomes, Mitochondria, Bacteria and Neurons. Bioelectrochemistry 2021, 137, 107673.
- Khailova, L.S.; Vygodina, T.V.; Lomakina, G.Y.; Kotova, E.A.; Antonenko, Y.N. Bicarbonate Suppresses Mitochondrial Membrane Depolarization Induced by Conventional Uncouplers. Biochem. Biophys. Res. Commun. 2020, 530, 29–34.
- Starkov, A.A.; Dedukhova, V.I.; Skulachev, V.P. 6-Ketocholestanol Abolishes the Effect of the Most Potent Uncouplers of Oxidative Phosphorylation in Mitochondria. FEBS Lett. 1994, 355, 305–308.
- Rousset, S.; Alves-Guerra, M.-C.; Mozo, J.; Miroux, B.; Cassard-Doulcier, A.-M.; Bouillaud, F.; Ricquier, D. The Biology of Mitochondrial Uncoupling Proteins. Diabetes 2004, 53, S130–S135.
- Plotnikov, E.Y.; Zorov, D.B. Pros and Cons of Use of Mitochondria-Targeted Antioxidants. Antioxidants 2019, 8, 316.
- Winslow, R.M. Oxygen: The Poison Is in the Dose. Transfusion 2013, 53, 424–437.
- Lyons, T.W.; Reinhard, C.T.; Planavsky, N.J. The Rise of Oxygen in Earth’s Early Ocean and Atmosphere. Nature 2014, 506, 307–315.
- Jones, D.P. Intracellular Diffusion Gradients of O2 and ATP. Am. J. Physiol. Cell Physiol. 1986, 250, C663–C675.
- Näpänkangas, J.P.; Liimatta, E.V.; Joensuu, P.; Bergmann, U.; Ylitalo, K.; Hassinen, I.E. Superoxide Production during Ischemia-Reperfusion in the Perfused Rat Heart: A Comparison of Two Methods of Measurement. J. Mol. Cell Cardiol. 2012, 53, 906–915.
- Waypa, G.B.; Schumacker, P.T. O2 Sensing in Hypoxic Pulmonary Vasoconstriction: The Mitochondrial Door Re-Opens. Respir. Physiol. Neurobiol. 2002, 132, 81–91.
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High Protonic Potential Actuates a Mechanism of Production of Reactive Oxygen Species in Mitochondria. FEBS Lett. 1997, 416, 15–18.
- Starkov, A.A.; Fiskum, G. Regulation of Brain Mitochondrial H2O2 Production by Membrane Potential and NAD(P)H Redox State. J. Neurochem. 2003, 86, 1101–1107.
- Skulachev, V.P. Anion Carriers in Fatty Acid-Mediated Physiological Uncoupling. J. Bioenerg. Biomembr. 1999, 31, 431–445.
- Severin, F.F.; Severina, I.I.; Antonenko, Y.N.; Rokitskaya, T.I.; Cherepanov, D.A.; Mokhova, E.N.; Vyssokikh, M.Y.; Pustovidko, A.V.; Markova, O.V.; Yaguzhinsky, L.S.; et al. Penetrating Cation/Fatty Acid Anion Pair as a Mitochondria-Targeted Protonophore. Proc. Natl. Acad. Sci. USA 2010, 107, 663–668.
- Trendeleva, T.A.; Sukhanova, E.I.; Rogov, A.G.; Zvyagilskaya, R.A.; Seveina, I.I.; Ilyasova, T.M.; Cherepanov, D.A.; Skulachev, V.P. Role of Charge Screening and Delocalization for Lipophilic Cation Permeability of Model and Mitochondrial Membranes. Mitochondrion 2013, 13, 500–506.
- Schrauwen, P.; Walder, K.; Ravussin, E. Human Uncoupling Proteins and Obesity. Obes. Res. 1999, 7, 97–105.
- Chen, S.-Y.; Beretta, M.; Alexopoulos, S.J.; Shah, D.P.; Olzomer, E.M.; Hargett, S.R.; Childress, E.S.; Salamoun, J.M.; Aleksovska, I.; Roseblade, A.; et al. Mitochondrial Uncoupler SHC517 Reverses Obesity in Mice without Affecting Food Intake. Metabolism 2021, 117, 154724.
- Axelrod, C.L.; King, W.T.; Davuluri, G.; Noland, R.C.; Hall, J.; Hull, M.; Dantas, W.S.; Zunica, E.R.; Alexopoulos, S.J.; Hoehn, K.L.; et al. BAM15-Mediated Mitochondrial Uncoupling Protects against Obesity and Improves Glycemic Control. EMBO Mol. Med. 2020, 12, e12088.
- Tainter, M.L.; Stockton, A.B.; Cutting, W.C. Dinitrophenol in the treatment of obesity: Final report. J. Am. Med. Assoc. 1935, 105, 332–337.
- Ruiz-Ramírez, A.; López-Acosta, O.; Barrios-Maya, M.A.; El-Hafidi, M. Cell Death and Heart Failure in Obesity: Role of Uncoupling Proteins. Oxid. Med. Cell. Longev. 2016, 2016, e9340654.
- Schlagowski, A.I.; Singh, F.; Charles, A.L.; Gali Ramamoorthy, T.; Favret, F.; Piquard, F.; Geny, B.; Zoll, J. Mitochondrial Uncoupling Reduces Exercise Capacity despite Several Skeletal Muscle Metabolic Adaptations. J. Appl. Physiol. 2014, 116, 364–375.
- Itami, N.; Shiratsuki, S.; Shirasuna, K.; Kuwayama, T.; Iwata, H. Mitochondrial Biogenesis and Degradation Are Induced by CCCP Treatment of Porcine Oocytes. Reproduction 2015, 150, 97–104.
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124.
- De Pauw, A.; Demine, S.; Tejerina, S.; Dieu, M.; Delaive, E.; Kel, A.; Renard, P.; Raes, M.; Arnould, T. Mild Mitochondrial Uncoupling Does Not Affect Mitochondrial Biogenesis but Downregulates Pyruvate Carboxylase in Adipocytes: Role for Triglyceride Content Reduction. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1123–E1141.
- Kansaku, K.; Takeo, S.; Itami, N.; Kin, A.; Shirasuna, K.; Kuwayama, T.; Iwata, H. Maternal Aging Affects Oocyte Resilience to Carbonyl Cyanide-m-Chlorophenylhydrazone -Induced Mitochondrial Dysfunction in Cows. PLoS ONE 2017, 12, e0188099.
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial Membrane Potential. Anal. Biochem. 2018, 552, 50–59.
- Hay, R.; Böhni, P.; Gasser, S. How Mitochondria Import Proteins. Biochim. Biophys. Acta 1984, 779, 65–87.
- Pfanner, N.; Tropschug, M.; Neupert, W. Mitochondrial Protein Import: Nucleoside Triphosphates Are Involved in Conferring Import-Competence to Precursors. Cell 1987, 49, 815–823.
- Neupert, W. Protein Import into Mitochondria. Annu. Rev. Biochem. 1997, 66, 863–917.
- Truscott, K.N.; Pfanner, N.; Voos, W. Transport of proteins into mitochondria. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2001; pp. 81–136. ISBN 978-3-540-44510-4.
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial Membrane Potential Regulates PINK1 Import and Proteolytic Destabilization by PARL. J. Cell Biol. 2010, 191, 933–942.
- Zorov, D.B.; Popkov, V.A.; Zorova, L.D.; Vorobjev, I.A.; Pevzner, I.B.; Silachev, D.N.; Zorov, S.D.; Jankauskas, S.S.; Babenko, V.A.; Plotnikov, E.Y. Mitochondrial Aging: Is There a Mitochondrial Clock? J. Gerontol. Biol. Sci. Med. Sci. 2017, 72, 1171–1179.
- Zorov, D.B.; Vorobjev, I.A.; Popkov, V.A.; Babenko, V.A.; Zorova, L.D.; Pevzner, I.B.; Silachev, D.N.; Zorov, S.D.; Andrianova, N.V.; Plotnikov, E.Y. Lessons from the Discovery of Mitochondrial Fragmentation (Fission): A Review and Update. Cells 2019, 8, 175.
- Gunter, K.K.; Gunter, T.E. Transport of Calcium by Mitochondria. J. Bioenerg. Biomembr. 1994, 26, 471–485.
- Gunter, T.E.; Pfeiffer, D.R. Mechanisms by Which Mitochondria Transport Calcium. Am. J. Physiol. Cell Physiol. 1990, 258, C755–C786.
- Duchen, M.R. Mitochondria and Calcium: From Cell Signalling to Cell Death. J. Physiol. 2000, 529, 57–68.
- Cortassa, S.; Juhaszova, M.; Aon, M.A.; Zorov, D.B.; Sollott, S.J. Mitochondrial Ca2+, Redox Environment and ROS Emission in Heart Failure: Two Sides of the Same Coin? J. Mol. Cell. Cardiol. 2021, 151, 113–125.
- Maragos, W.F.; Korde, A.S. Mitochondrial Uncoupling as a Potential Therapeutic Target in Acute Central Nervous System Injury. J. Neurochem. 2004, 91, 257–262.
- Stout, A.K.; Raphael, H.M.; Kanterewicz, B.I.; Klann, E.; Reynolds, I.J. Glutamate-Induced Neuron Death Requires Mitochondrial Calcium Uptake. Nat. Neurosci. 1998, 1, 366–373.
- Khodorov, B.I.; Storozhevykh, T.P.; Surin, A.M.; Yuryavichyus, A.I.; Sorokina, E.G.; Borodin, A.V.; Vinskaya, N.P.; Khaspekov, L.G.; Pinelis, V.G. The Leading Role of Mitochondrial Depolarization in the Mechanism of Glutamate-Induced Disruptions in Ca2+ Homeostasis. Neurosci. Behav. Physiol. 2002, 32, 541–547.
- Korde, A.S.; Sullivan, P.G.; Maragos, W.F. The Uncoupling Agent 2,4-Dinitrophenol Improves Mitochondrial Homeostasis Following Striatal Quinolinic Acid Injections. J. Neurotrauma 2005, 22, 1142–1149.
- Gohel, D.; Singh, R. Mitohormesis; Potential Implications in Neurodegenerative Diseases. Mitochondrion 2021, 56, 40–46.
- Tapia, P.C. Sublethal Mitochondrial Stress with an Attendant Stoichiometric Augmentation of Reactive Oxygen Species May Precipitate Many of the Beneficial Alterations in Cellular Physiology Produced by Caloric Restriction, Intermittent Fasting, Exercise and Dietary Phytonutrients: “Mitohormesis” for Health and Vitality. Med. Hypotheses 2006, 66, 832–843.
- Jankauskas, S.S.; Silachev, D.N.; Andrianova, N.V.; Pevzner, I.B.; Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Zorov, D.B. Aged Kidney: Can We Protect It? Autophagy, Mitochondria and Mechanisms of Ischemic Preconditioning. Cell Cycle 2018, 17, 1291–1309.
- Silachev, D.N.; Plotnikov, E.Y.; Pevzner, I.B.; Zorova, L.D.; Babenko, V.A.; Zorov, S.D.; Popkov, V.A.; Jankauskas, S.S.; Zinchenko, V.P.; Sukhikh, G.T.; et al. The Mitochondrion as a Key Regulator of Ischaemic Tolerance and Injury. HeartLung Circ. 2014, 23, 897–904.
- Murry, C.E.; Richard, V.J.; Jennings, R.B.; Reimer, K.A. Myocardial Protection Is Lost Before Contractile Function Recovers from Ischemic Preconditioning. Am. J. Physiol. Heart Circ. Physiol. 1991, 260, H796–H804.
- Liu, Y.; Kato, H.; Nakata, N.; Kogure, K. Protection of Rat Hippocampus against Ischemic Neuronal Damage by Pretreatment with Sublethal Ischemia. Brain Res. 1992, 586, 121–124.
- Pashkovskaya, A.A.; Vazdar, M.; Zimmermann, L.; Jovanovic, O.; Pohl, P.; Pohl, E.E. Mechanism of Long-Chain Free Fatty Acid Protonation at the Membrane-Water Interface. Biophys. J. 2018, 114, 2142–2151.
- Jezek, P. Fatty Acid Interaction with Mitochondrial Uncoupling Proteins. J. Bioenerg. Biomembr. 1999, 31, 457–466.
- Andreyev, A.Y.; Bondareva, T.O.; Dedukhova, V.I.; Mokhova, E.N.; Skulachev, V.P.; Tsofina, L.M.; Volkov, N.I.; Vygodina, T.V. The ATP/ADP-Antiporter Is Involved in the Uncoupling Effect of Fatty Acids on Mitochondria. Eur. J. Biochem. 1989, 182, 585–592.
- Wieckowski, M.R.; Wojtczak, L. Involvement of the Dicarboxylate Carrier in the Protonophoric Action of Long-Chain Fatty Acids in Mitochondria. Biochem. Biophys. Res. Commun. 1997, 232, 414–417.
- Hodges, J.M.; Gutenstein, M.; Marx, W. Thyroxine and Yeast Metabolism: Uncoupling of Phosphorylation. Arch. Biochem. Biophys. 1963, 101, 429–435.
- Klemperer, H.G. The Uncoupling of Oxidative Phosphorylation in Rat-Liver Mitochondria by Thyroxine, Triiodothyronine and Related Substances. Biochem. J. 1955, 60, 122–128.
- Hoch, F.L.; Lipmann, F. The Uncoupling of Respiration and Phosphorylation by Thyroid Hormones. Proc. Natl. Acad. Sci. USA 1954, 40, 909–921.
- Yau, W.W.; Singh, B.K.; Lesmana, R.; Zhou, J.; Sinha, R.A.; Wong, K.A.; Wu, Y.; Bay, B.-H.; Sugii, S.; Sun, L.; et al. Thyroid Hormone (T3) Stimulates Brown Adipose Tissue Activation via Mitochondrial Biogenesis and MTOR-Mediated Mitophagy. Autophagy 2019, 15, 131–150.
- Martinez-Lopez, N.; Garcia-Macia, M.; Sahu, S.; Athonvarangkul, D.; Liebling, E.; Merlo, P.; Cecconi, F.; Schwartz, G.J.; Singh, R. Autophagy in the CNS and Periphery Coordinate Lipophagy and Lipolysis in the Brown Adipose Tissue and Liver. Cell Metab. 2016, 23, 113–127.
- Konrad, C.; Kawamata, H.; Bredvik, K.G.; Arreguin, A.J.; Cajamarca, S.A.; Hupf, J.C.; Ravits, J.M.; Miller, T.M.; Maragakis, N.J.; Hales, C.M.; et al. Fibroblast Bioenergetics to Classify Amyotrophic Lateral Sclerosis Patients. Mol. Neurodegener. 2017, 12, 76.
- Perera, N.D.; Turner, B.J. AMPK Signalling and Defective Energy Metabolism in Amyotrophic Lateral Sclerosis. Neurochem. Res. 2016, 41, 544–553.
- Desport, J.C.; Preux, P.M.; Magy, L.; Boirie, Y.; Vallat, J.M.; Beaufrère, B.; Couratier, P. Factors Correlated with Hypermetabolism in Patients with Amyotrophic Lateral Sclerosis. Am. J. Clin. Nutr. 2001, 74, 328–334.
- Sarnat, H.B.; Flores-Sarnat, L.; Hader, W.; Bello-Espinosa, L. Mitochondrial “Hypermetabolic” Neurons in Paediatric Epileptic Foci. Can. J. Neurol. Sci 2011, 38, 909–917.
- Herrero, A.; Barja, G. ADP-Regulation of Mitochondrial Free Radical Production Is Different with Complex I- or Complex II-Linked Substrates: Implications for the Exercise Paradox and Brain Hypermetabolism. J. Bioenerg. Biomembr. 1997, 29, 241–249.
- O’Shaughnessy, C.T.; Rothwell, N.J.; Shrewsbury-Gee, J. Sympathetically Mediated Hypermetabolic Response to Cerebral Ischemia in the Rat. Can. J. Physiol. Pharm. 1990, 68, 1334–1337.
- Shabalina, I.G.; Nedergaard, J. Mitochondrial (‘mild’) Uncoupling and ROS Production: Physiologically Relevant or Not? Biochem. Soc. Trans. 2011, 39, 1305–1309.

