 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Fatemeh Mirzadeh Azad | + 2012 word(s) | 2012 | 2021-07-20 08:09:27 | | | |
| 2 | Nora Tang | + 255 word(s) | 2267 | 2021-08-23 09:50:13 | | | | |
| 3 | Nora Tang | + 255 word(s) | 2267 | 2021-08-23 09:51:06 | | |
Video Upload Options
One of the major challenges in cancer treatments is the dynamic adaptation of tumor cells to cancer therapies. In this regard, tumor cells can modify their response to environmental cues without altering their DNA sequence. This cell plasticity enables cells to undergo morphological and functional changes, for example, during the process of tumour metastasis or when acquiring resistance to cancer therapies. Central to cell plasticity, are the dynamic changes in gene expression that are controlled by a set of molecular switches called enhancers. Enhancers are DNA elements that determine when, where and to what extent genes should be switched on and off. Thus, defects in enhancer function can disrupt the gene expression program and can lead to tumour formation. Here, we review how enhancers control the activity of cancer-associated genes and how defects in these regulatory elements contribute to cell plasticity in cancer. Understanding enhancer (de)regulation can provide new strategies for modulating cell plasticity in tumour cells and can open new research avenues for cancer therapy.
1. The Epigenetic Foundation of Tumour Plasticity
One of the processes that underlies tumour heterogeneity is the cellular differentiation of tumour cells that harbour a stem cell capacity. These cancer stem cells (CSCs) generate committed cells with a spectrum of phenotypes, that all share the same genetic background [1][2], making CSCs one of the main culprits of tumour heterogeneity [3][4]. Recent studies suggest that stemness can be considered as a “biological state” in which cells can enter or exit, indicating a robust cell plasticity within tumours [5][6]. Here, tumour cells can gain stem-like or differentiated phenotype depending on the intrinsic genetic triggers or the external environmental cues. For example, in luminal breast tumours, in which the majority of cells have epidermal characteristics, a small fraction of cells expresses the mesenchymal/basal markers (e.g., CD44) and show resemblance with the normal mammary stem cells. Of note, a homogenous population of luminal tumour cells, sorted based on low levels of CD44(CD44 low ), can regenerate the tumour bulk that contains CD44 high cells (constituting 10% of the tumour bulk). Thus, the luminal breast tumour cells can undergo de-differentiation to obtain a more basal CD44 high phenotype, suggesting a strong plasticity of cells within the tumour [6].
In addition to phenotypic variation, our understanding of the extent of tumour heterogeneity has tremendously increased by using technologies that reveal features of tumour cells at the single cell level. One such approach is based on investigating the gene expression profiles by high-resolution single cell RNA-seq (scRNA-seq) analysis [7]. For example, a recent study of a BRCA1-null model of breast cancer indicates that tumour cells with a similar genetic background can cluster into different subpopulations that have distinct gene expression profiles. In this regard, the upregulation of cell cycle regulators (e.g., BIRC5 , TYMS and MKI67 ) is only observed in a cluster of highly proliferative cells and not in the cell cluster that exhibits a progenitor-like phenotype (with the expression of the basal cell markers such as KRT14 , IGFBP5 , WNT10A ). These observations suggest that cancer therapies that target cell proliferation may be less effective in eradicating the quiescent progenitor-like populations in these tumours [8][9][10].
The exact sequence of events that leads to enhancer activation is not well established; however, pioneering factors and lineage defining TFs (LDTFs) can access the condensed chromatin regions and can act as the initial step for enhancer activation. These TFs recruit the epigenetic modifiers, e.g., the histone acetyl transferases CBP/P300 and/or the mono methyl transferases MLL3/MLL4, to deposit H3K27ac and H3K4me1 at enhancers. These active enhancer marks are then recognised by epigenetic readers such as BRD4 that recruit transcription coactivator, the Mediator complex and the RPOL2 transcription machinery to maintain the expression of target genes [11][12].
The activity of enhancers can be also regulated by DNA methylation that is deposited at cytosine residues and, based on the genomic location and the co-occurrence of other epigenetic marks, can have a positive or negative impact on transcription. Generally, CpGs methylation at promoters and enhancers is followed by inactivating histone methylation marks and the formation of a condensed chromatin state [13], whereas gene-body methylation shows a positive correlation with transcription [14]. The global hypomethylation, e.g., via mutation in DNMTs, can potentiate the ectopic expression of oncogenes [15]. For example, studies in lymphoma indicate that the hypomethylated fraction of a genome usually harbours genes and CREs that are related to proliferation, differentiation, and negative regulators of P53 pathway [16]. However, an elevated level of DNMT can also contribute to tumourigenesis through the inactivation of tumour suppressor genes [17]. In breast cancer, for instance, the upregulation of DNMTs is necessary for tumour progression; here, the cancer stem cell subpopulation relies on DNMT1 to hypermethylate and suppress ISL1 that functions as a negative regulator of self-renewal in mammary stem cells and plays a tumour suppressor role in breast cancer [18][19].
2. Enhancer Dynamics and Tumour Plasticity:
Switching between enhancers in different developmental states is another strategy that cells use to tailor their gene expression profiles. In haematopoietic stem and progenitor cells, alternative elements that engage in enhancer switching have different compositions of TF binding sites that include various differentiation regulators (such as MYB, FLI1, LMO2, and RUNX1) and signalling-dependent TFs. This unique array of TF-binding sites and the differential expression of TFs determines the state-specificity and the level of enhancer activity. Interestingly, the deregulation of these TFs is often detected in transformed cells, leading to enhancer switching and the usage of elements with more oncogenic impacts. In line with this, the expression of GATA2 and MYC in haematopoietic malignancies is controlled by the re-establishment of a series of enhancers that normally function in HSCs, further highlighting the contribution of stem-state-specific enhancers to cancer [20][21][22].
EMT is one of the main mechanisms fuelling the tumour plasticity and heterogeneity and entails dynamic changes in transcriptional and epigenetic landscapes ( Figure 1 ) [23][24]. Studying EMT in normal and transformed cells indicates dependency on global epigenetic reprograming, that starts with extracellular cytokine or the induction of EMT master regulators (e.g., TWIST and SNAIL ). DNA methylation is one of the waves of epigenetic changes that is pivotal for the EMT cell state transition, as suppressing DNMTs (e.g., by chemical inhibition of their functions) impairs EMT, even when the EMT-inducing signal persists [24][25][26]. The reprogramming of DNA methylome starts shortly after EMT induction (e.g., by TGFB) and lasts until the cells commit to a mesenchymal state. In transitioning cells, hyper methylation occurs at CpGs containing regulatory regions that associate with epithelial identity and cell cycle progression, resulting in chromatin condensation at these sites. Histone modifications are another layer of epigenetic regulations that dynamically change in response to an elevated level of EMT master regulators such as SNAIL [27][28][29]. SNAIL mainly functions as a transcription repressor and modulates the loss of H3K27Ac and H3K4Me3, as well as the enrichment of H3K27me3 by recruiting corresponding chromatin modifiers to the promoter of epithelial markers. However, later on in the transition, SNAIL induces positive histone marks (H3K4Me3 and H3K4Me1) on the promoter of mesenchymal genes to support the mesenchymal commitment [27].
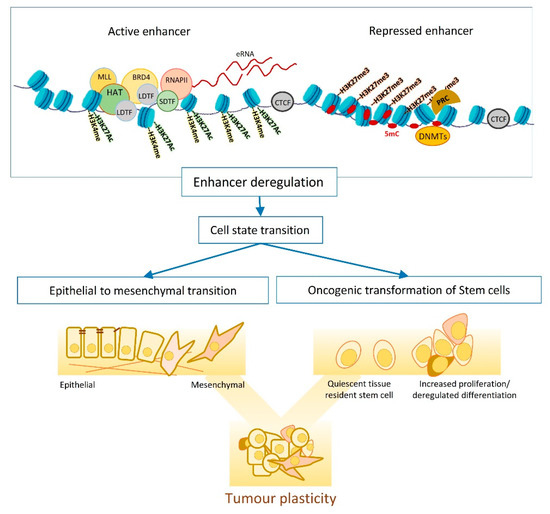
Among different extracellular signals, TGFB is one of the most potent inducers of EMT that triggers a global epigenetic reprogramming in target cells. Several signalling cascades function downstream of TGFB to transmit the signal to target genes. For example, in mammary epithelial cells, ERK signalling plays a crucial role downstream of TGFB to regulate H3K27ac deposition at enhancers. In this regard, enhancers activated by TGFB are highly enriched for TFs such as GABPA, JUN, RUNX1, and ATF3 that function downstream of ERK signalling and have prominent roles in EMT. Furthermore, when ERK-signalling is inhibited, cells treated with TGFB fail to express early EMT regulators such as HMGA2, ITGA2, and TGFBR1 due to a lack of H3K27ac deposition at corresponding enhancers. Thus, ERK-signalling is necessary for acquiring H3K27 acetylation at enhancers and the induction of the EMT process [30][31]. The TGFB-induced EMT is not always conveyed through canonical TFs (e.g., SNALI or ZEB1). In alveolar carcinoma cells, an unconventional trio of ETS2, HNF4A, JUNB show overexpression in the E/M hybrid phase of EMT and are necessary for the formation of TGFB-induced super enhancers. These super enhancers control the expression of mesenchymal markers such as FOXP1 and CDH2 in transitioning cells. The synergic effect of ETS2, HNF4A, JUNB is crucial for enhancer activity as their suppression impairs the EMT process through loss of enhancer activity [32].
EMT regulation can be cell-type specific, and the downstream epigenetic changes can vary, dependent on the EMT stimuli. For example, EMT induction in renal, alveolar and breast immortalised cells reveals that the changes in histone methylation (at H3K27, H3K4 and H3K9) is cell-type specific. This difference in epigenetic regulation is also observed when different EMT stimuli such as TGFB/TNFa or EGF are used. This context-specific epigenetic regulation is associated with different transcriptional outputs. For instance, TGFB treatment causes the downregulation of E-cadherin in all investigated cell lines, whereas the gain of mesenchymal markers such as Vimentin is detected in some cells. Nevertheless, a fraction of regulatory elements is similarly decorated with histone marks in all conditions, regardless of the source of EMT induction. Interestingly, these universal EMT elements control genes that function in extracellular matrix degradation (e.g., ADAM and MMP9 ) rather than EMT-related transcription factors [33][34].
3. Cancer-Related Genetic Variations at Enhancers
Somatic structural variants may also disrupt the boundaries of insulated genomic domains and can lead to deregulated gene expression ( Figure 2 C). Most enhancer–promoter interactions occur within topologically associating domains (TADs) [35] that insulate the regulatory activity of enhancers from neighbouring domains. In this regard, structural variants can lead to TADs fusion, the duplication of TAD-boundaries or complex rearrangements, such as TAD inversions. The PCWAG found that structural variants affecting TAD boundaries occur in 5.0%, 8.5%, and 12.8% of all deletions, inversions, and duplications, respectively. Such examples have been observed in a TAD boundary deletion near the WNT4 locus in lymphoma, and the SLC22A2 locus in breast cancer. However, PCWAG results indicate that structural variation at TAD boundaries do not strongly affect the expression of nearby genes; only in 14% of the cases, deletion in a TAD boundary results in the significant expression change of nearby genes [36].
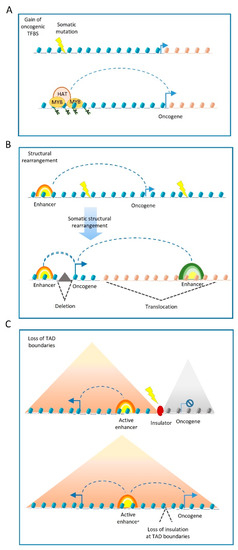
It is important to note that enhancers operate as platforms for the epigenetic machinery. Thus, in addition to genetic variations in cis-regulatory elements, mutations in enhancer-associated proteins can also influence cell state, tumour formation and progression, via altered epi-decoration of enhancers [37][38].
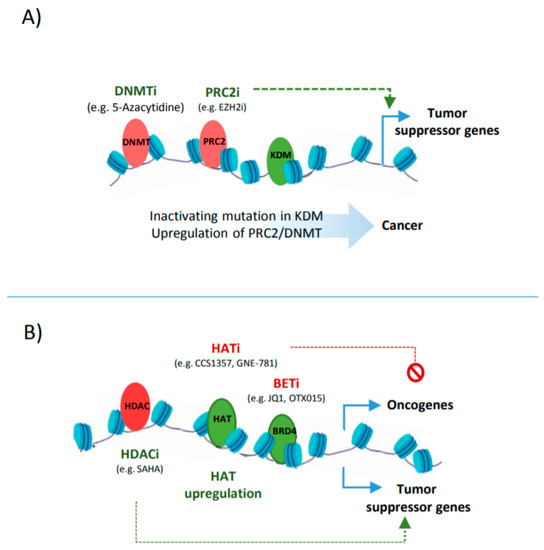
The other side of balancing the histone acetylation is controlled by histone deacetylases such as HDAC1. A multi-omics study in liposarcoma shows that HDAC1 is mutated in 8.5% of primary tumours. Mechanistically, HDAC1 inactivation leads to the deregulated expression of lineage-defining TFs such as C/EBPα, enhancing the undifferentiated state of tumour cells [39]. In another case, CRC cells that carry a frame-shift mutation at the HDAC2 gene become refractory to anti-proliferative drugs. This loss of HDAC2 correlates with increased histone acetylation at regulatory elements that control various pathways involved in cell proliferation [40][41].
In addition to histone methylation, aberrant histone acetylation is also a major influencer of suppressor genes and oncogenes expression ( Figure 3 B). Therefore, enhancer reprogramming via targeting histone acetylation provides a promising therapeutic strategy in cancer. One such strategy is based on the upregulation of HDACs or inhibiting the activity of HATs, leading to a loss of the acetylation at CREs associated with oncogenes [42]. Such examples include the use of small molecules, such as Comp 5, that induces SIRT1 catalytic activity, resulting in the deacetylation of H3 in glioma or using HAT inhibitors such as CCS1357 for targeting P300/CBP in prostate cancer, leading to the downregulation of AR and MYC [43][44]. The combined inhibition of epi-factors, e.g., P300/CBP (by GNE-781) and BRD4 (by OTX015), have been also applied for decommissioning oncogenic enhancers (e.g., MYC enhancers), and can increase the anti-tumour efficacy [45][46]. Targeting tumour-associated super enhancers also provides a promising epigenetic therapy, as addiction to oncogenic enhancers can be observed in many tumours [47].
References
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406.
- Von Loga, K.; Woolston, A.; Punta, M.; Barber, L.J.; Griffiths, B.; Semiannikova, M.; Spain, G.; Challoner, B.; Fenwick, K.; Simon, R.; et al. Extreme intratumour heterogeneity and driver evolution in mismatch repair deficient gastro-oesophageal cancer. Nat. Commun. 2020, 11, 1–14.
- Carpenter, R.; Sirkisoon, S.; Zhu, D.; Rimkus, T.; Harrison, A.; Anderson, A.; Paw, I.; Qasem, S.; Xing, F.; Liu, Y.; et al. Combined inhibition of AKT and HSF1 suppresses breast cancer stem cells and tumor growth. Oncotarget 2017, 8, 73947–73963.
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.N.S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5+ human colon cancer stem cells. Nat. Cell Biol. 2017, 545, 187–192.
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.K.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1–16.
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955.
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e18.
- Fu, N.; Rios, A.C.; Pal, B.; Law, C.W.; Jamieson, P.; Liu, R.; Vaillant, F.; Jackling, F.; Liu, K.H.; Smyth, G.K.; et al. Identification of quiescent and spatially restricted mammary stem cells that are hormone responsive. Nat. Cell Biol. 2017, 19, 164–176.
- Yeo, S.K.; Zhu, X.; Okamoto, T.; Hao, M.; Wang, C.; Lu, P.; Lu, L.J.; Guan, J.-L. Single-cell RNA-sequencing reveals distinct patterns of cell state heterogeneity in mouse models of breast cancer. eLife 2020, 9.
- Russnes, H.G.; Navin, N.; Hicks, J.; Borresen-Dale, A.-L. Insight into the heterogeneity of breast cancer through next-generation sequencing. J. Clin. Investig. 2011, 121, 3810–3818.
- Lee, J.-E.; Park, Y.-K.; Park, S.; Jang, Y.; Waring, N.; Dey, A.; Ozato, K.; Lai, B.; Peng, W.; Ge, K. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun. 2017, 8, 1–12.
- Mullen, A.C.; Orlando, D.A.; Newman, J.J.; Lovén, J.; Kumar, R.M.; Bilodeau, S.; Reddy, J.; Guenther, M.G.; DeKoter, R.; Young, R.A. Master Transcription Factors Determine Cell-Type-Specific Responses to TGF-β Signaling. Cell 2011, 147, 565–576.
- Wang, X.; Paucek, R.D.; Gooding, A.R.; Brown, Z.Z.; Ge, E.J.; Muir, T.W.; Cech, T.R. Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nat. Struct. Mol. Biol. 2017, 24, 1028–1038.
- Arechederra, M.; Daian, F.; Yim, A.; Bazai, S.K.; Richelme, S.; Dono, R.; Saurin, A.J.; Habermann, B.H.; Maina, F. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat. Commun. 2018, 9, 1–16.
- Sheaffer, K.L.; Elliott, E.N.; Kaestner, K.H. DNA Hypomethylation Contributes to Genomic Instability and Intestinal Cancer Initiation. Cancer Prev. Res. 2016, 9, 534–546.
- Kushwaha, G.; Dozmorov, M.; Wren, J.D.; Qiu, J.; Shi, H.; Xu, D. Hypomethylation coordinates antagonistically with hypermethylation in cancer development: A case study of leukemia. Hum. Genom. 2016, 10, 83–102.
- García-Martínez, A.; Sottile, J.; Sánchez-Tejada, L.; Fajardo, C.; Cámara, R.; Lamas, C.; Barberá, V.M.; Picó, A. DNA Methylation of Tumor Suppressor Genes in Pituitary Neuroendocrine Tumors. J. Clin. Endocrinol. Metab. 2019, 104, 1272–1282.
- Gao, X.; Sheng, Y.; Yang, J.; Wang, C.; Zhang, R.; Zhu, Y.; Zhang, Z.; Zhang, K.; Yan, S.; Sun, H.; et al. Osteopontin alters DNA methylation through up-regulating DNMT1 and sensitizes CD133+/CD44+ cancer stem cells to 5 azacytidine in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 1–14.
- Pathania, R.; Ramachandran, S.; Elangovan, S.; Padia, R.; Yang, P.; Cinghu, S.; Veeranan-Karmegam, R.; Arjunan, P.; Gnana-Prakasam, J.P.; Sadanand, F.; et al. DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat. Commun. 2015, 6, 1–11.
- Benetatos, L.; Benetatou, A.; Vartholomatos, G. Enhancers and MYC interplay in hematopoiesis. J. Mol. Med. 2020, 98, 471–481.
- Bahr, C.; Von Paleske, L.; Uslu, V.V.; Remeseiro, S.; Takayama, N.; Ng, S.W.; Murison, A.; Langenfeld, K.; Petretich, M.; Scognamiglio, R.; et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nat. Cell Biol. 2018, 553, 515–520.
- Mehta, C.; Johnson, K.D.; Gao, X.; Ong, I.M.; Katsumura, K.R.; McIver, S.C.; Ranheim, E.A.; Bresnick, E.H. Integrating Enhancer Mechanisms to Establish a Hierarchical Blood Development Program. Cell Rep. 2017, 20, 2966–2979.
- Bhatia, S.; Monkman, J.; Blick, T.; Pinto, C.; Waltham, M.; Nagaraj, S.H.; Thompson, E.W. Interrogation of Phenotypic Plasticity between Epithelial and Mesenchymal States in Breast Cancer. J. Clin. Med. 2019, 8, 893.
- Choi, S.K.; Pandiyan, K.; Eun, J.W.; Yang, X.; Hong, S.H.; Nam, S.W.; Jones, P.A.; Liang, G.; You, J.S. Epigenetic landscape change analysis during human EMT sheds light on a key EMT mediator TRIM29. Oncotarget 2017, 8, 98322–98335.
- Galle, E.; Thienpont, B.; Cappuyns, S.; Venken, T.; Busschaert, P.; Van Haele, M.; Van Cutsem, E.; Roskams, T.; Van Pelt, J.; Verslype, C.; et al. DNA methylation-driven EMT is a common mechanism of resistance to various therapeutic agents in cancer. Clin. Epigenetics 2020, 12, 1–19.
- Cardenas, H.; Vieth, E.; Lee, J.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. TGF-β induces global changes in DNA methylation during the epithelial-to-mesenchymal transition in ovarian cancer cells. Epigenetics 2014, 9, 1461–1472.
- Javaid, S.; Zhang, J.; Anderssen, E.; Black, J.; Wittner, B.S.; Tajima, K.; Ting, D.; Smolen, G.A.; Zubrowski, M.; Desai, R.; et al. Dynamic Chromatin Modification Sustains Epithelial-Mesenchymal Transition following Inducible Expression of Snail-1. Cell Rep. 2013, 5, 1679–1689.
- Battistelli, C.; Cicchini, C.; Santangelo, L.; Tramontano, A.; Grassi, L.; Gonzalez, F.J.; De Nonno, V.; Grassi, G.; Amicone, L.; Tripodi, M. The Snail repressor recruits EZH2 to specific genomic sites through the enrollment of the lncRNA HOTAIR in epithelial-to-mesenchymal transition. Oncogene 2016, 36, 942–955.
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail Mediates E-Cadherin Repression by the Recruitment of the Sin3A/Histone Deacetylase 1 (HDAC1)/HDAC2 Complex. Mol. Cell. Biol. 2004, 24, 306–319.
- Navandar, M.; Garding, A.; Sahu, S.K.; Pataskar, A.; Schick, S.; Tiwari, V.K. ERK signalling modulates epigenome to drive epithelial to mesenchymal transition. Oncotarget 2017, 8, 29269–29281.
- Shin, S.; Buel, G.R.; Nagiec, M.J.; Han, M.-J.; Roux, P.P.; Blenis, J.; Yoon, S.-O. ERK2 regulates epithelial-to-mesenchymal plasticity through DOCK10-dependent Rac1/FoxO1 activation. Proc. Natl. Acad. Sci. USA 2019, 116, 2967–2976.
- Chang, H.; Liu, Y.; Xue, M.; Liu, H.; Du, S.; Zhang, L.; Wang, P. Synergistic action of master transcription factors controls epithelial-to-mesenchymal transition. Nucleic Acids Res. 2016, 44, 2514–2527.
- Peixoto, P.; Etcheverry, A.; Aubry, M.; Missey, A.; Lachat, C.; Perrard, J.; Hendrick, E.; Delage-Mourroux, R.; Mosser, J.; Borg, C.; et al. EMT is associated with an epigenetic signature of ECM remodeling genes. Cell Death Dis. 2019, 10, 1–17.
- Vidakovic, A.T.; Rueda, O.M.; Vervoort, S.J.; Batra, A.S.; Goldgraben, M.; Uribe-Lewis, S.; Greenwood, W.; Coffer, P.J.; Bruna, A.; Caldas, C. Context-Specific Effects of TGF-β/SMAD3 in Cancer Are Modulated by the Epigenome. Cell Rep. 2015, 13, 2480–2490.
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680.
- Zhang, Y.; PCAWG Transcriptome Working Group; Chen, F.; Fonseca, N.A.; He, Y.; Fujita, M.; Nakagawa, H.; Zhang, Z.; Brazma, A.; Creighton, C.J.; et al. High-coverage whole-genome analysis of 1220 cancers reveals hundreds of genes deregulated by rearrangement-mediated cis-regulatory alterations. Nat. Commun. 2020, 11, 1–14.
- De Matos, M.R.; Posa, I.; Carvalho, F.; Morais, V.A.; Grosso, A.R.; De Almeida, S.F. A Systematic Pan-Cancer Analysis of Genetic Heterogeneity Reveals Associations with Epigenetic Modifiers. Cancers 2019, 11, 391.
- Gagliardi, A.; Dugué, P.-A.; Nøst, T.H.; Southey, M.C.; Buchanan, D.D.; Schmidt, D.F.; Makalic, E.; Hodge, A.M.; English, D.R.; Doo, N.W.; et al. Stochastic Epigenetic Mutations Are Associated with Risk of Breast Cancer, Lung Cancer, and Mature B-cell Neoplasms. Cancer Epidemiol. Biomark. Prev. 2020, 29, 2026–2037.
- Taylor, B.S.; DeCarolis, P.L.; Angeles, C.V.; Brenet, F.; Schultz, N.; Antonescu, C.R.; Scandura, J.; Sander, C.; Viale, A.J.; Socci, N.D.; et al. Frequent Alterations and Epigenetic Silencing of Differentiation Pathway Genes in Structurally Rearranged Liposarcomas. Cancer Discov. 2011, 1, 587–597.
- Ropero, S.; Fraga, M.; Ballestar, E.; Hamelin, R.; Yamamoto, H.; Boix-Chornet, M.; Caballero, R.T.; Alaminos, M.; Setien, F.; Paz, M.F.; et al. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat. Genet. 2006, 38, 566–569.
- Wang, P.; Wang, Z.; Liu, J. Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 2020, 19, 1–21.
- Cole, K.E.; Dowling, D.P.; Boone, M.A.; Phillips, A.J.; Christianson, D.W. Structural Basis of the Antiproliferative Activity of Largazole, a Depsipeptide Inhibitor of the Histone Deacetylases. J. Am. Chem. Soc. 2011, 133, 12474–12477.
- Brooks, N.; Pegg, N.; Worthington, J.; Young, B.; Prosser, A.; Lane, J.; Taddei, D.; Schiewer, M.J.; Gordon, N.; Knudsen, K.E. A novel small molecule inhibitor of p300/CBP for the treatment of castration-resistant prostate cancer: Preclinical evaluation. J. Clin. Oncol. 2017, 35, 168.
- Yao, Z.-Q.; Zhang, X.; Zhen, Y.; He, X.-Y.; Zhao, S.; Li, X.-F.; Yang, B.; Gao, F.; Guo, F.-Y.; Fu, L.; et al. A novel small-molecule activator of Sirtuin-1 induces autophagic cell death/mitophagy as a potential therapeutic strategy in glioblastoma. Cell Death Dis. 2018, 9, 1–14.
- Morrison-Smith, C.D.; Knox, T.M.; Filic, I.; Soroko, K.M.; Eschle, B.K.; Wilkens, M.K.; Gokhale, P.C.; Giles, F.; Griffin, A.; Brown, B.; et al. Combined Targeting of the BRD4–NUT–p300 Axis in NUT Midline Carcinoma by Dual Selective Bromodomain Inhibitor, NEO2734. Mol. Cancer Ther. 2020, 19, 1406–1414.
- Wiese, M.; Hamdan, F.H.; Kubiak, K.; Diederichs, C.; Gielen, G.H.; Nussbaumer, G.; Carcaboso, A.M.; Hulleman, E.; Johnsen, S.A.; Kramm, C.M. Combined treatment with CBP and BET inhibitors reverses inadvertent activation of detrimental super enhancer programs in DIPG cells. Cell Death Dis. 2020, 11, 1–13.
- Gryder, B.E.; Yohe, M.E.; Chou, H.-C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3–FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov. 2017, 7, 884–899.


