 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Petra Dunkel | + 2645 word(s) | 2645 | 2021-07-08 04:03:41 |
Video Upload Options
Lack of selectivity and toxic side effects of cancer chemotherapy is a major drawback for designing clinical treatment regimes. To allow more potent and selective therapeutic interventions, the use of light for selective on-site activation of anticancer compounds is actively studied. Besides already established photodynamic therapy (PDT), two light-activated anticancer approaches are pursued, both however still in the experimental phase: the use of irreversibly activatable photoremovable protecting groups (“photocages”) and reversibly activatable photoswitches. Despite its immense potential, light activation brings many novel challenges to the already complex drug development process, however the first in vivo results confirm the feasibility of the approach.
1. Introduction
2. Irreversible Activation with Light: Photoremovable Protecting Groups (“Photocages”) for Antitumor Applications
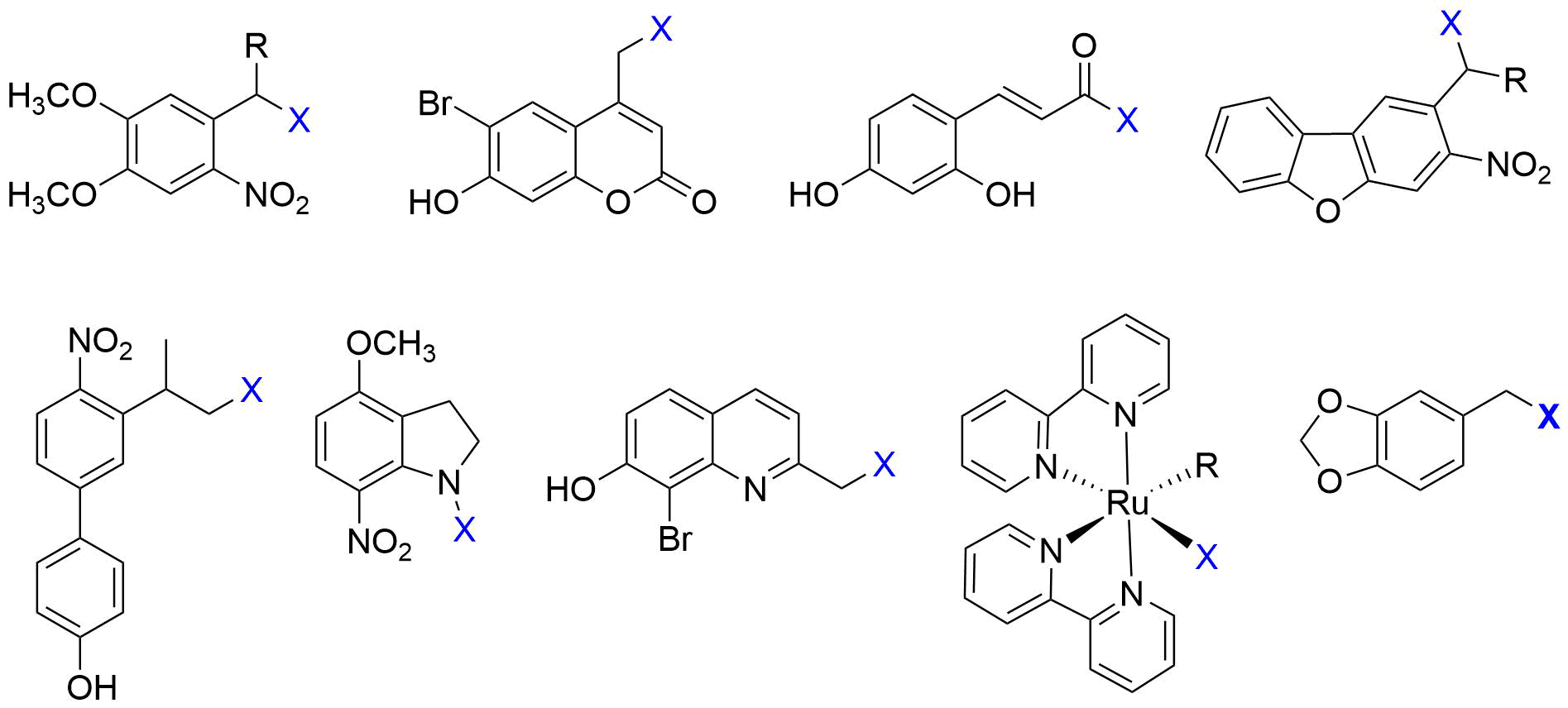
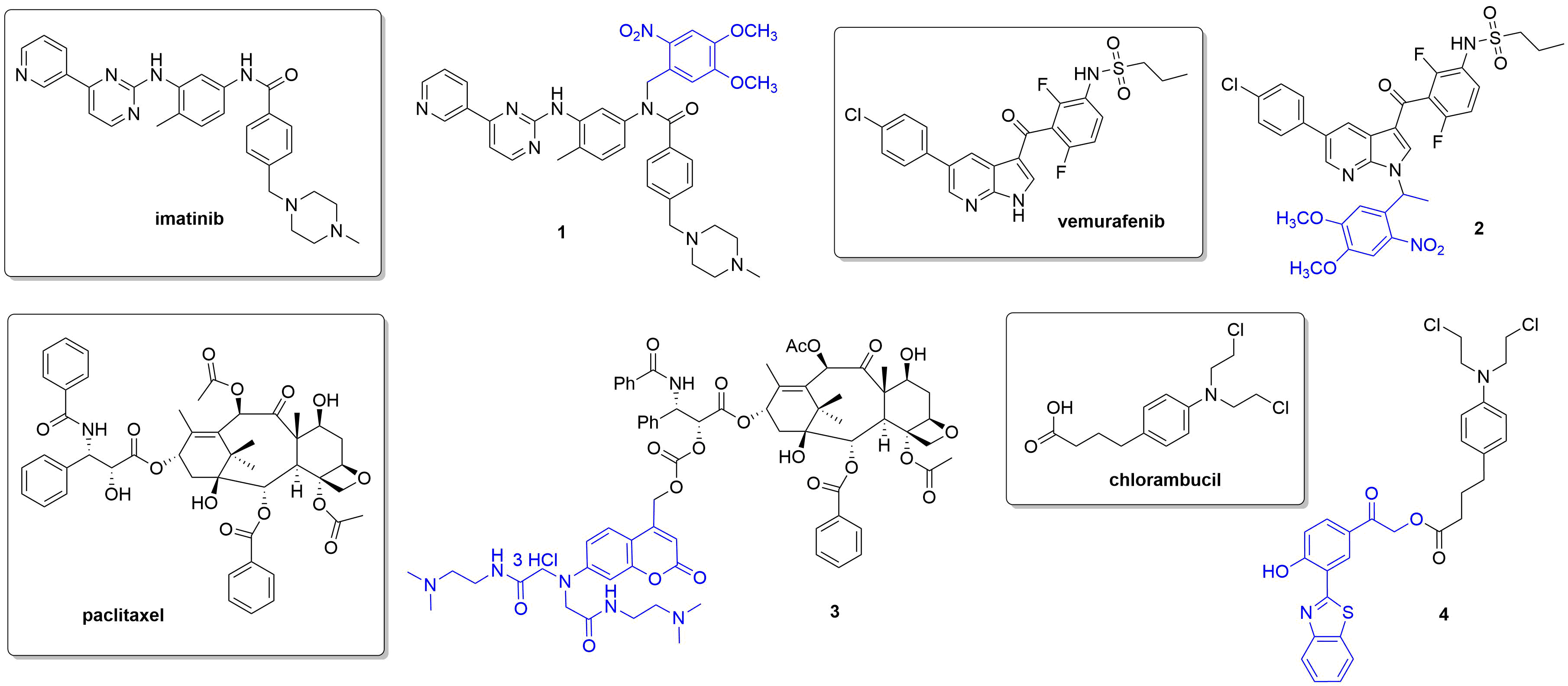
Figure 3. Examples of PPG-based photoactivatable analogues of approved anticancer drugs, with the structures of the parent drugs in the boxes and the PPG units indicated in blue in the photoactivatable versions, respectively (1[15], 2[16], 3[20], 4[36]).
3. Reversible Activation with Light: Photoremovable Protecting Groups (“Photocages”) for Antitumor Applications
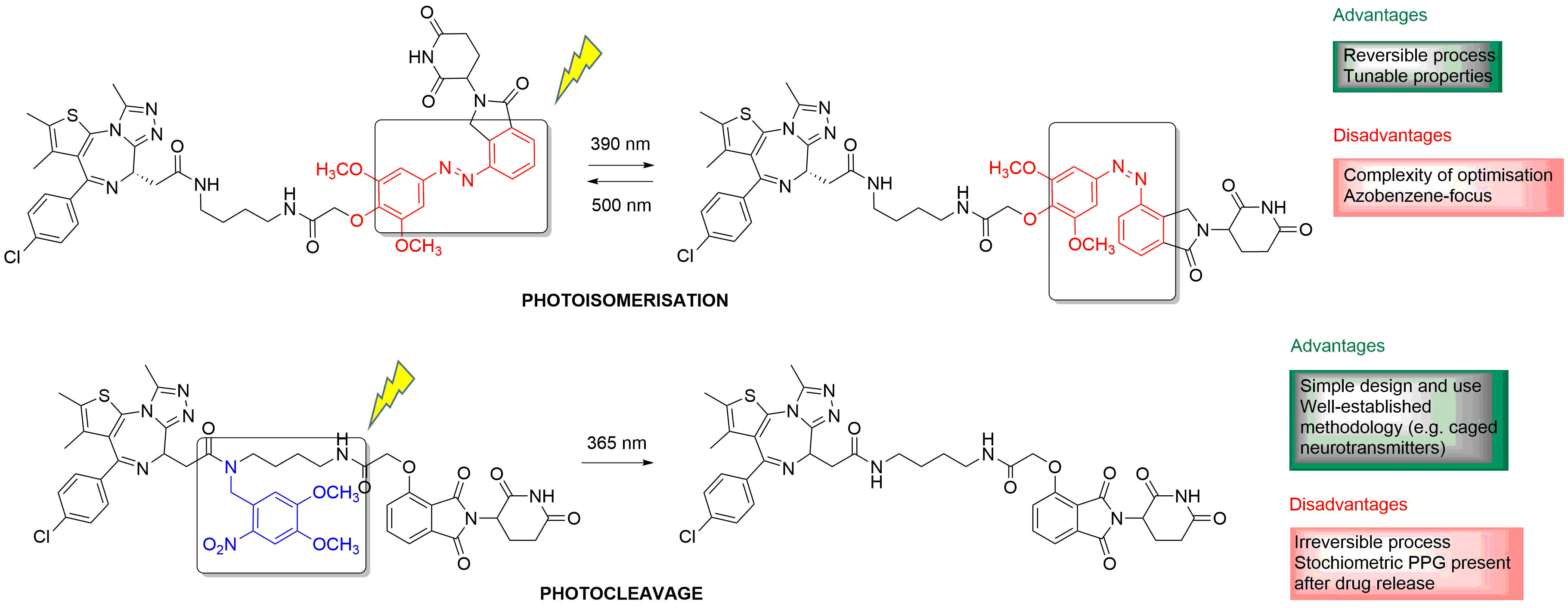
As the concept of photocages, that of photoswitches also dates back to several decades[39], however particularly the recent years have seen a surge of activity in the field (referred to as photopharmacology)[40][41]. The application of photoswitchable pharmacological agents is based on their two (or more) reversibly interconvertible isomeric forms that allow significant steric changes upon photoisomerization and, consequently, different pharmacological activity (Figure 4). The photoisomerization could proceed via two principal routes, trans→cis (E→Z) isomerization (e.g., azobenzenes and their heteroaromatic analogs, indigos, hemithioindigos, stilbenes, hydrazones, and iminothioindoxyls) or 6π electrocyclization of a triene system (e.g., diarylethenes)[42]. Of further types, spiropyrans, fulgides, and donor-acceptor Stenhouse adducts were exploited[43].
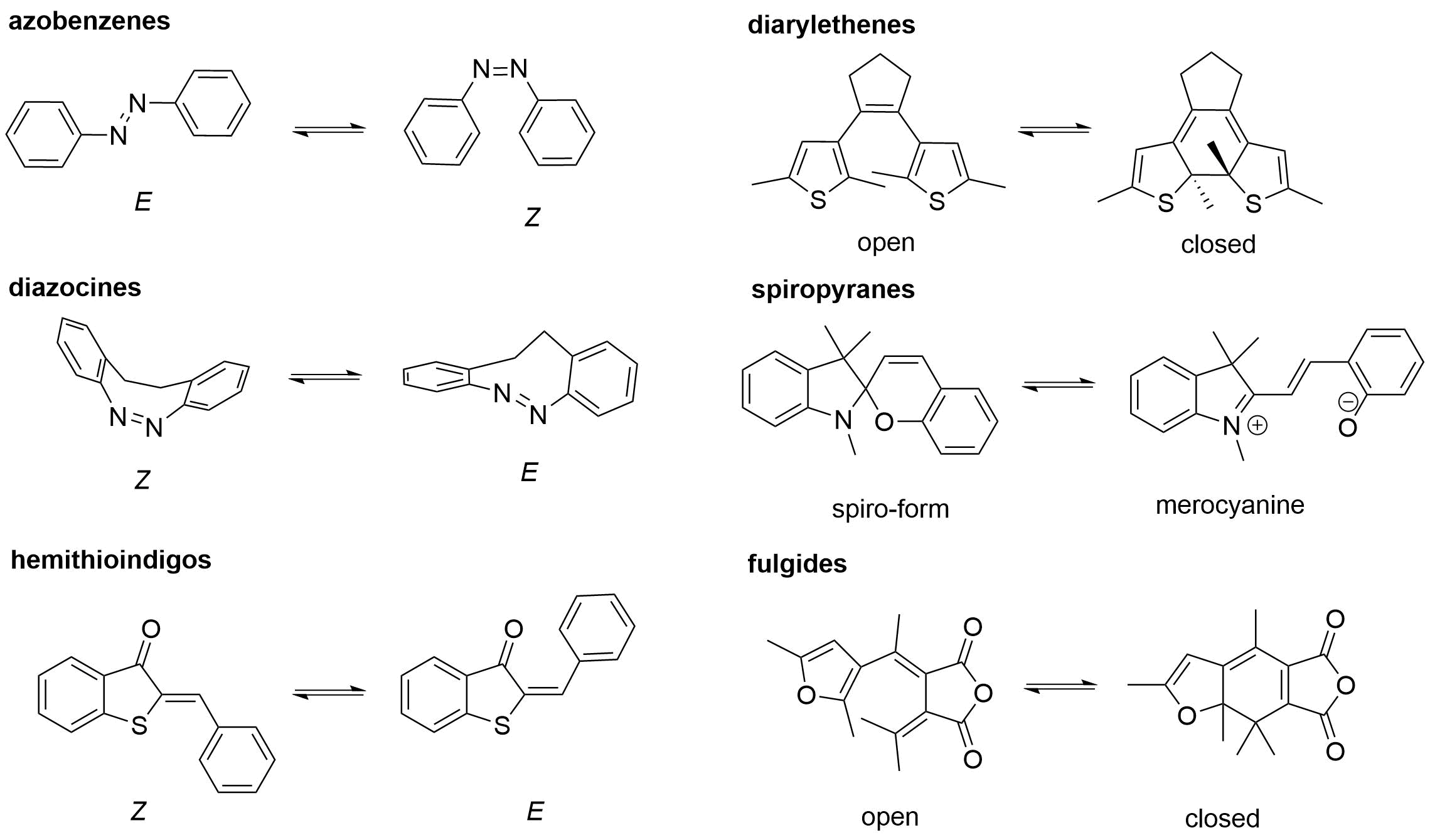
Figure 4. Representative examples of photoswitch scaffolds[42].
Although the reversible activation of photoswitches confers many advantages, it also adds further layers of complexity. Selective irradiation of the isomers is feasible in the case of appropriate band separation, and for further applications in living systems, the activation wavelengths should ideally be in the biological window. Furthermore, the probes should ideally have high photofatigue resistance, rapid isomerization kinetics, a photostationary state (PSS) sufficiently enriched in the active isomer, as well as a half-life of the metastable state in line with the planned application. Depending on the target and the probe’s structure, different scenarios are possible (unlike the typically one-way turn-on activation of photocages). Both the thermodynamically more stable (dark) or the less stable (obtained upon light activation) form could have higher bioactivity. Typically, the sought-for scenario is where the active form is obtained upon irradiation (i.e., a turn-on activity); thereby, administration problems could be avoided or background activity resulting from incomplete photoisomerization. The active form could isomerize back to the inactive isomer thermally (T-type photoswitches, thermally reversible) or upon light irradiation (P-type photoswitches, photochemically reversible), therefore upon deactivation a more localized effect could be obtained. Besides the photophysical and photochemical properties optimised for the intended application, the novel probes should also comply with the criteria of pharmacological/therapeutic applications, as lack of ground state toxicity, feasible hydrolytic solubility and stability, or resistance toward reduction in biological media (e.g., by glutathione)[44]. Obtaining a balanced activity profile and at the same time appropriate photoactivation could be particularly challenging. The general design strategy for reversibly photoactivatable drug molecules is either to add a photoswitchable (often arylazo → “azo-extension”) tag to the pharmacophore of the parent structure or to incorporate a (often arylazo → “azologization”) photoswitchable unit into the pharmacophore. The latter strategy could be expected to alter less the overall structure, therefore having less impact on the pharmacokinetic and pharmacodynamic properties of the parent drug. Evidently, for both approaches besides a clinically validated target, detailed SAR and structural information are necessary of the target and the parent drug (family). From a practical point of view, synthetic accessibility of the novel probes, availability of straightforward methods for the desired structural modifications should also be duly considered.
Exploiting the available structural information, photoswitchable analogues of several approved anticancer drugs were prepared and tested (e.g. axitinib[45][46], vorinostat[47], methotrexate[48], paclitaxel[49], bortezomib[50][51]) (Figure 5). The choice of the parent drug is often guided by structural elements present in the molecule allowing photoisomerization themselves or upon modification (e.g. an activity difference was recorded already for the E and Z isomers of combretastatin A4 and a number of photoswitchable analogs were designed subsequently by replacing the central C=C bond with an N=N unit[52][53][54][55][56]). As for PPG-based probes, also for photoswitches a number of anticancer pharmacological classes were addressed (several classes were the target of both approaches), including kinase inhibitors (e.g.[45][46]), epigenetic modulators (e.g.[47][57]), antimetabolites[48], microtubule-targeting agents (e.g.[53][54]), proteasome inhibitors[50][51] or PROTACs (e.g.[58][59]). Of the pharmacological classes studied for both approaches[60][61], PROTACs emerged recently in the forefront of interest as a tool for inducing targeted protein degradation. PROTACs are bifunctional molecules that could facilitate binding between an E3 ligase and a chosen protein, thereby inducing protein degradation via the ubiquitin-proteasome pathway. With PROTACs formerly not druggable proteins could be addressed as well, including key players of oncogenic processes. The modular design of PROTACs also facilitates incorporating the respective photoactivatable structural elements.
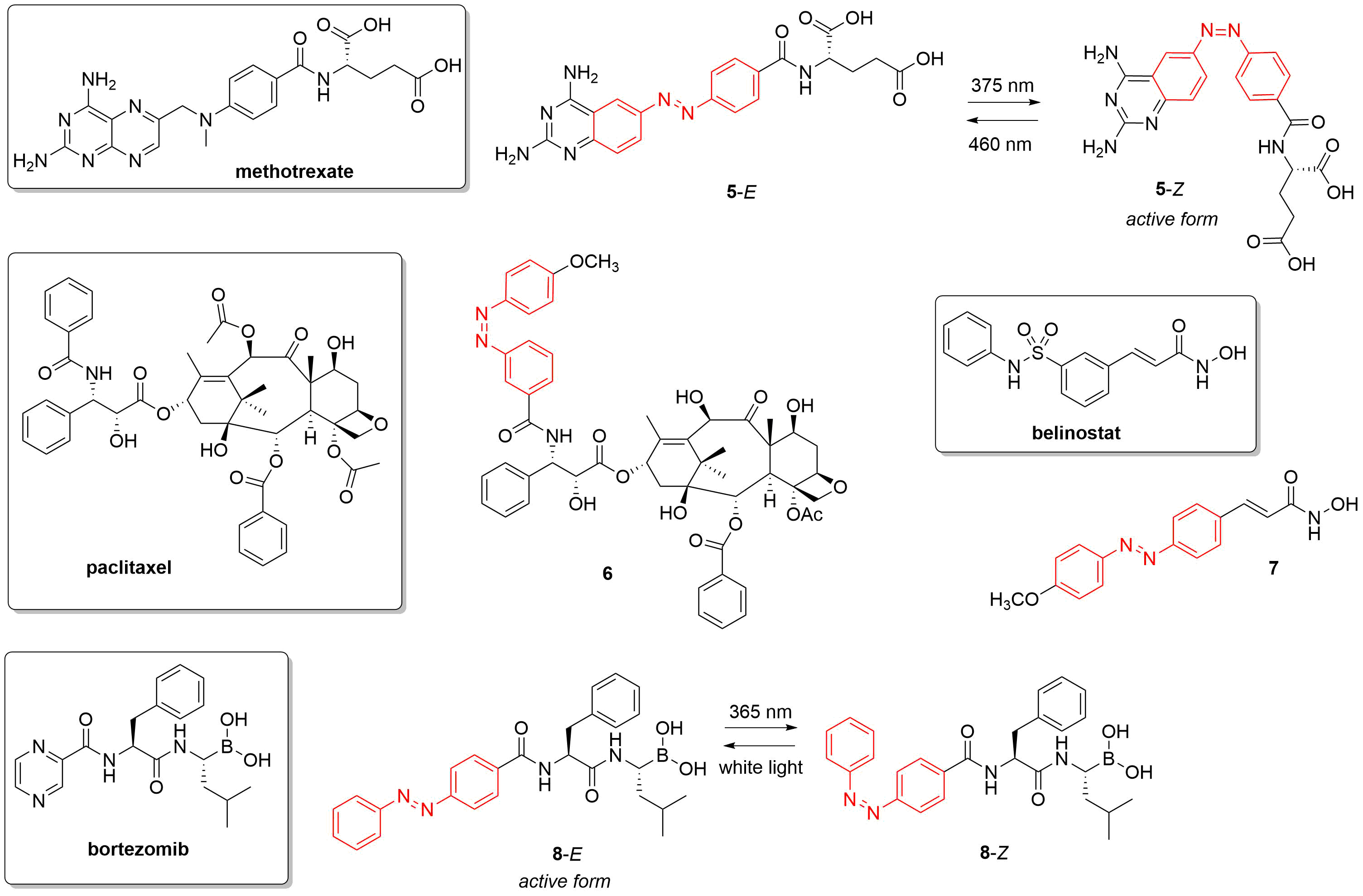
Figure 5. Examples of photoswitchable analogues of approved anticancer drugs, with the structures of the parent drugs in the boxes and the photoswitch units indicated in red in the photoactivatable versions, respectively (5[48], 6[49], 7[47], 8[50]).
4. Challenges and Future Perspectives for Photoactivatable Anticancer Agents
As any clinical drugs, reversibly or irreversibly photoactivatable anticancer agents should comply with a plethora of pharmacokinetic, and pharmacodynamic criteria besides having photophysical and photochemical characteristics optimised for the intended use. Many of the challenges that externally addressable drug delivery systems face (e.g. tissue penetration, delivery to and retention at the active site, control over activation signal in vivo, materials and instrumentation complexity, available translational models and safety[62]) are also relevant for the field of light-responsive (small) drug molecules (Figure 6).

Figure 6. Challenges and future perspectives for photoactivatable anticancer agents.
A specific challenge is the wavelength required for the photoactivation and consequently the tissue penetration of the light trigger. In this respect, probes operating at higher wavelengths (red, infrared) or ideally in the biological window (λ = 650–1450 nm)[63] would be advantageous, and many efforts have been dedicated to developing such photoactivatable units. Optimisation of the photoactivation step should not impair however the other properties of the probes and a balanced profile should be attained. For instance, in the field of azobenzene photoswitches, longer wavelength absorption generally leads also to faster thermal relaxation; moreover, the relative stabilities of the Z and E isomers could be altered as well. A typical approach for shifting the absorption toward higher wavelengths is to prepare push-pull systems with appropriately positioned electron-donating and electron-withdrawing groups. Not all structural modifications leading to higher wavelength absorption are feasible however, if they lead to a construct beyond the drug-like size or to a high number of rotatable bonds. For obtaining small-molecule azo-photoswitches with red-shifted absorption and slow (s/min) thermal relaxation in water, altering e.g. judiciously the substitution pattern might offer a straightforward route[64]. Diazocines (bridged azobenzenes) offer complete switching in both directions, a red-shifted absorbance, and a thermal relaxation rate in the minutes range[65][66]. However, due to the cyclic structure, the Z isomer is the more stable one, which is typically the less sought-for option. In the field of photocages, for targeting the biological window examples of probes operating at long wavelengths, e.g., boron dipyrromethene (BODIPY) or heptamethine cyanine derivatives, could be cited[67][68][69][70][71].
To circumvent light-irradiation wavelength issues, using specifically designed probes both photoisomerization and photocleavage might profit from two-photon absorption. Two-photon absorption (TPA) is a nonlinear optical process, necessitating in practice the use of femtosecond pulsed laser systems. The simultaneous absorption of two low-energy photons (occurring only at high light intensity) leads to photoreaction. Especially in the field of photocages, a wide variety of two-photon activatable probes are already available, and work is continuously in progress for further derivatives with improved photophysical and photochemical profiles[72][73]. Next to modifications of the chromophore itself, specific formulations could also allow harnessing longer wavelength light, e.g., the use of upconverting nano-particles or alternative sources of light, as the Cherenkov radiation (i.e., a design based on internal co-localization of the light source and the photoactive agent)[74].
A major bottleneck at present is, that typically reports disclosing photoactivatable probes are based on in vitro assays (proof-of-concept studies). For future therapeutic applications, it would be highly needed to gain a better insight into how these systems could be operated under in vivo conditions. The most studied in vivo application of photoactivatable agents so far is vision restoration, a logical choice regarding the site of activation[75]. However, it demonstrates the applicability of the concept per se. For targeting tumors in deeper tissues, further obstacles still need to be overcome.
Regarding the future scope and potential of light-activatable agents, besides releasing the active (form of the) drug, further functions could be envisaged as well. Notably, photoresponsive probes could also offer in situ monitoring of the drug release (i.e., rational dosimetry). Typically for photocages, release or formation of a fluorescent species is a general approach for real-time monitoring of the photorelease. Particularly when moving toward in vivo systems, getting quantitative information on free drug concentration has tremendous practical importance. Particularly in the realm of nanodevices, light-triggered action could also be complemented with imaging modalities (i.e., theranostic construct designs)[76][77][78].
On-target activity requirements for photocages and photoswitches differ substantially. Photocages rely absolutely on the activity of the released compound, i.e., native ligand, which is usually a clinically used drug. Photocaging approach, however, also offers a possibility to use more potent (toxic) compounds, which cannot be used directly due to side effects or unsuitable physicochemical and pharmacokinetic properties. Additionally, the introduction of PPGs enables tuning drug-likeness of the prodrug (solubility, permeability, etc.). The on-target activity of photoswitches is a considerably more demanding task. Usually, photoswitchable moieties are relatively big and might substantially impair intrinsic binding. Nevertheless, several literature examples demonstrate the feasibility of the task. At this point, photoswitchable PROTACs are specific and successful examples offering a general approach to locating photoswitchable moieties as linker units or on E3 ligase recruiting ligand and not interfering with the protein recruiting part.
As the discovery of anticancer compounds (or novel APIs themselves) is a highly demanding task, adding another level of complexity raises several further concerns, such as regulatory issues, different pharmacokinetic properties of the isomers, the dependence of the photochemical properties on the (biological) environment to name just a few. Currently, we are in the phase where the concept of light-responsive anticancer agents is being developed and is focused on optimizing compound properties, while biological activity is mainly validated with cancer cell line assays (with the exception of a yet small number of in vivo animal studies[18][79]). Rapid development and constantly improved constructs are leading researchers into the stage that photoresponsive compounds will be evaluated in animal models, and proposed improvements in anticancer therapy will be thus validated.
References
- Michael J. Hassett; A. James O'malley; Juliana R. Pakes; Joseph P. Newhouse; Craig C. Earle; Frequency and Cost of Chemotherapy-Related Serious Adverse Effects in a Population Sample of Women With Breast Cancer. JNCI Journal of the National Cancer Institute 2006, 98, 1108-1117, 10.1093/jnci/djj305.
- Vladimir P. Torchilin; Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nature Reviews Drug Discovery 2014, 13, 813-827, 10.1038/nrd4333.
- Y. Lee; D.H. Thompson; Stimuli-responsive liposomes for drug delivery. WIREs Nanomedicine and Nanobiotechnology 2017, 9, e1450, 10.1002/wnan.1450.
- Simona Mura; Julien Nicolas; Patrick Couvreur; Stimuli-responsive nanocarriers for drug delivery. Nature Materials 2013, 12, 991-1003, 10.1038/nmat3776.
- Zheng Huang; A Review of Progress in Clinical Photodynamic Therapy. Technology in Cancer Research & Treatment 2005, 4, 283-293, 10.1177/153303460500400308.
- Chase Linsley; Benjamin M Wu; Recent advances in light-responsive on-demand drug-delivery systems. Therapeutic Delivery 2017, 8, 89-107, 10.4155/tde-2016-0060.
- Mahdi Karimi; Parham Sahandi Zangabad; Soodeh Baghaee-Ravari; Mehdi Ghazadeh; Hamid Mirshekari; Michael R. Hamblin; Smart Nanostructures for Cargo Delivery: Uncaging and Activating by Light. Journal of the American Chemical Society 2017, 139, 4584-4610, 10.1021/jacs.6b08313.
- Petr Klán; Tomáš Šolomek; Christian G. Bochet; Aurélien Blanc; Richard Givens; Marina Rubina; Vladimir Popik; Alexey Kostikov; Jakob Wirz; Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy. Chemical Reviews 2012, 113, 119-191, 10.1021/cr300177k.
- Aurélien Blanc; Christian G. Bochet; Wavelength-Controlled Orthogonal Photolysis of Protecting Groups. The Journal of Organic Chemistry 2002, 67, 5567-5577, 10.1021/jo025837m.
- Srinivas Kantevari; Masanori Matsuzaki; Yuya Kanemoto; Haruo Kasai; Graham C.R. Ellis-Davies; Two-color, two-photon uncaging of glutamate and GABA.. Nature Chemical Biology 2009, 7, 123-125, 10.1038/nmeth.1413.
- Mickel J. Hansen; Willem A. Velema; Michael M. Lerch; Wiktor Szymanski; Ben L. Feringa; Wavelength-selective cleavage of photoprotecting groups: strategies and applications in dynamic systems. Chemical Society Reviews 2015, 44, 3358-3377, 10.1039/c5cs00118h.
- Hsien-Ming Lee; Daniel Larson; David S. Lawrence; Illuminating the Chemistry of Life: Design, Synthesis, and Applications of “Caged” and Related Photoresponsive Compounds. ACS Chemical Biology 2009, 4, 409-427, 10.1021/cb900036s.
- Balázs Chiovini; Dénes Pálfi; Myrtill Majoros; Gábor Juhász; Gergely Szalay; Gergely Katona; Milán Szőri; Orsolya Frigyesi; Csilla Lukácsné Haveland; Gábor Szabó; et al.Ferenc ErdélyiZoltán MátéZoltán SzadaiMiklós MadarászMiklós DékányImre G. CsizmadiaErvin KovácsBalázs RózsaZoltán Mucsi Theoretical Design, Synthesis, and In Vitro Neurobiological Applications of a Highly Efficient Two-Photon Caged GABA Validated on an Epileptic Case. ACS Omega 2021, 6, 15029-15045, 10.1021/acsomega.1c01164.
- Jarkko Rautio; Hanna Kumpulainen; Tycho Heimbach; Reza Oliyai; Dooman Oh; Tomi Järvinen; Jouko Savolainen; Prodrugs: design and clinical applications. Nature Reviews Drug Discovery 2008, 7, 255-270, 10.1038/nrd2468.
- Melanie Zindler; Boris Pinchuk; Christian Renn; Rebecca Horbert; Alexander Döbber; Christian Peifer; Design, Synthesis, and Characterization of a Photoactivatable Caged Prodrug of Imatinib. ChemMedChem 2015, 10, 1335-1338, 10.1002/cmdc.201500163.
- Rebecca Horbert; Boris Pinchuk; Paul Davies; Dario Alessi; Christian Peifer; Photoactivatable Prodrugs of Antimelanoma Agent Vemurafenib. ACS Chemical Biology 2015, 10, 2099-2107, 10.1021/acschembio.5b00174.
- Stuart Ibsen; Eran Zahavy; Wolf Wrasdilo; Michael Berns; Michael Chan; Sadik Esener; A Novel Doxorubicin Prodrug with Controllable Photolysis Activation for Cancer Chemotherapy. Pharmaceutical Research 2010, 27, 1848-1860, 10.1007/s11095-010-0183-x.
- Stuart Ibsen; Eran Zahavy; Wolf Wrasidlo; Tomoko Hayashi; John Norton; Yongxuan Su; Stephen Adams; Sadik Esener; Localized in vivo activation of a photoactivatable doxorubicin prodrug in deep tumor tissue.. Photochemistry and Photobiology 2013, 89, 698-708, 10.1111/php.12045.
- Mariusz Skwarczynski; Mayo Noguchi; Shun Hirota; Youhei Sohma; Tooru Kimura; Yoshio Hayashi; Yoshiaki Kiso; Development of first photoresponsive prodrug of paclitaxel. Bioorganic & Medicinal Chemistry Letters 2006, 16, 4492-4496, 10.1016/j.bmcl.2006.06.030.
- Mayo Noguchi; Mariusz Skwarczynski; Halan Prakash; Shun Hirota; Tooru Kimura; Yoshio Hayashi; Yoshiaki Kiso; Development of novel water-soluble photocleavable protective group and its application for design of photoresponsive paclitaxel prodrugs. Bioorganic & Medicinal Chemistry 2008, 16, 5389-5397, 10.1016/j.bmc.2008.04.022.
- Amrita Paul; Angana Biswas; Sreyashi Sinha; Sk. Sheriff Shah; Manoranjan Bera; Mahitosh Mandal; N. D. Pradeep Singh; Push–Pull Stilbene: Visible Light Activated Photoremovable Protecting Group for Alcohols and Carboxylic Acids with Fluorescence Reporting Employed for Drug Delivery. Organic Letters 2019, 21, 2968-2972, 10.1021/acs.orglett.9b00124.
- S. Karthik; B. N. Prashanth Kumar; Moumita Gangopadhyay; Mahitosh Mandal; N. D. Pradeep Singh; A targeted, image-guided and dually locked photoresponsive drug delivery system. Journal of Materials Chemistry B 2014, 3, 728-732, 10.1039/c4tb01583e.
- Sarit S. Agasti; Ashley Laughney; Rainer H. Kohler; Ralph Weissleder; A photoactivatable drug-caged fluorophore conjugate allows direct quantification of intracellular drug transport.. Chemical Communications 2013, 49, 11050-11052, 10.1039/c3cc46089d.
- Jiaguo Li; Dian Xiao; Lianqi Liu; Fei Xie; Wei Li; Wei Sun; Xiaohong Yang; Xinbo Zhou; Design, Synthesis, and In Vitro Evaluation of the Photoactivatable Prodrug of the PARP Inhibitor Talazoparib. Molecules 2020, 25, 407, 10.3390/molecules25020407.
- Naoya Ieda; Sota Yamada; Mitsuyasu Kawaguchi; Naoki Miyata; Hidehiko Nakagawa; (7-Diethylaminocoumarin-4-yl)methyl ester of suberoylanilide hydroxamic acid as a caged inhibitor for photocontrol of histone deacetylase activity. Bioorganic & Medicinal Chemistry 2016, 24, 2789-2793, 10.1016/j.bmc.2016.04.042.
- Anna Leonidova; Cristina Mari; Christine Aebersold; Gilles Gasser; Selective Photorelease of an Organometallic-Containing Enzyme Inhibitor. Organometallics 2016, 35, 851-854, 10.1021/acs.organomet.6b00029.
- Tanmaya Joshi; Vanessa Pierroz; Cristina Mari; Lea Gemperle; Stefano Ferrari; Gilles Gasser; A Bis(dipyridophenazine)(2-(2-pyridyl)pyrimidine-4-carboxylic acid)ruthenium(II) Complex with Anticancer Action upon Photodeprotection. Angewandte Chemie International Edition 2014, 53, 2960-2963, 10.1002/anie.201309576.
- Katie L. Ciesienski; Lynne M. Hyman; Daniel T. Yang; Kathryn L. Haas; Marina G. Dickens; Robert J. Holbrook; Katherine J. Franz; A Photo-Caged Platinum(II) Complex That Increases Cytotoxicity upon Light Activation. European Journal of Inorganic Chemistry 2010, 2010, 2224-2228, 10.1002/ejic.201000098.
- Gang Xue; Kun Wang; Danli Zhou; Hanbing Zhong; Zhengying Pan; Light-Induced Protein Degradation with Photocaged PROTACs. Journal of the American Chemical Society 2019, 141, 18370-18374, 10.1021/jacs.9b06422.
- Cyrille Stephane Kounde; Maria Shchepinova; Charlie N. Saunders; Marcel X. Muelbaier; Mark D. Rackham; John D. Harling; Edward William Tate; A caged E3 ligase ligand for PROTAC-mediated protein degradation with light. Chemical Communications 2020, 56, 5532-5535, 10.1039/d0cc00523a.
- Mickel J. Hansen; Femke Feringa; Piermichele Kobauri; Wiktor Szymanski; René H. Medema; Ben L. Feringa; Photoactivation of MDM2 Inhibitors: Controlling Protein–Protein Interaction with Light. Journal of the American Chemical Society 2018, 140, 13136-13141, 10.1021/jacs.8b04870.
- Pamela T. Wong; Shengzhuang Tang; Jayme Cannon; Jhindan Mukherjee; Danielle Isham; Kristina Gam; Michael Payne; Sean A. Yanik; James R. Baker Jr.; Seok Ki Choi; et al. A Thioacetal Photocage Designed for Dual Release: Application in the Quantitation of Therapeutic Release by Synchronous Reporter Decaging. ChemBioChem 2016, 18, 126-135, 10.1002/cbic.201600494.
- Thomas A. Shell; Jennifer R. Shell; Zachary L. Rodgers; David S. Lawrence; Tunable Visible and Near-IR Photoactivation of Light-Responsive Compounds by Using Fluorophores as Light-Capturing Antennas. Angewandte Chemie International Edition 2013, 53, 875-878, 10.1002/anie.201308816.
- Radu A. Gropeanu; Hella Baumann; Sandra Ritz; Volker Mailänder; Thomas Surrey; Aránzazu Del Campo; Phototriggerable 2′,7-Caged Paclitaxel. PLOS ONE 2012, 7, e43657, 10.1371/journal.pone.0043657.
- Yarra Venkatesh; Y Rajesh; S. Karthik; A C Chetan; Mahitosh Mandal; Avijit Jana; N. D. Pradeep Singh; Photocaging of Single and Dual (Similar or Different) Carboxylic and Amino Acids by Acetyl Carbazole and its Application as Dual Drug Delivery in Cancer Therapy. The Journal of Organic Chemistry 2016, 81, 11168-11175, 10.1021/acs.joc.6b02152.
- Shrabani Barman; Sourav K. Mukhopadhyay; Sandipan Biswas; Surajit Nandi; Moumita Gangopadhyay; Satyahari Dey; Anakuthil Anoop; N. D. Pradeep Singh; A p -Hydroxyphenacyl-Benzothiazole-Chlorambucil Conjugate as a Real-Time-Monitoring Drug-Delivery System Assisted by Excited-State Intramolecular Proton Transfer. Angewandte Chemie International Edition 2016, 55, 4194-4198, 10.1002/anie.201508901.
- Andrew R. Hilgenbrink; Philip Low; Folate Receptor-Mediated Drug Targeting: From Therapeutics to Diagnostics. Journal of Pharmaceutical Sciences 2005, 94, 2135-2146, 10.1002/jps.20457.
- Martin Dcona; Jonathon E. Sheldon; Deboleena Mitra; Matthew C.T. Hartman; Light induced drug release from a folic acid-drug conjugate. Bioorganic & Medicinal Chemistry Letters 2016, 27, 466-469, 10.1016/j.bmcl.2016.12.036.
- H. Kaufman; S. M. Vratsanos; B. F. Erlanger; Photoregulation of an Enzymic Process by Means of a Light-Sensitive Ligand. Science 1968, 162, 1487-1489, 10.1126/science.162.3861.1487.
- Willem A. Velema; Wiktor Szymanski; Bernard Feringa; Photopharmacology: Beyond Proof of Principle. Journal of the American Chemical Society 2014, 136, 2178-2191, 10.1021/ja413063e.
- Wiktor Szymanski; John M. Beierle; Hans A. V. Kistemaker; Willem A. Velema; Ben L. Feringa; Reversible Photocontrol of Biological Systems by the Incorporation of Molecular Photoswitches. Chemical Reviews 2013, 113, 6114-6178, 10.1021/cr300179f.
- Ilse M. Welleman; Mark W. H. Hoorens; Ben L. Feringa; Hendrikus H. Boersma; Wiktor Szymański; Photoresponsive molecular tools for emerging applications of light in medicine. Chemical Science 2020, 11, 11672-11691, 10.1039/d0sc04187d.
- Luuk Kortekaas; Wesley R. Browne; The evolution of spiropyran: fundamentals and progress of an extraordinarily versatile photochrome. Chemical Society Reviews 2019, 48, 3406-3424, 10.1039/c9cs00203k.
- Ali Ryan; Azoreductases in drug metabolism. British Journal of Pharmacology 2016, 174, 2161-2173, 10.1111/bph.13571.
- Dorian Schmidt; Theo Rodat; Linda Heintze; Jantje Weber; Rebecca Horbert; Ulrich Girreser; Tim Raeker; Lara Bußmann; Malte Kriegs; Bernd Hartke; et al.Christian Peifer Axitinib: A Photoswitchable Approved Tyrosine Kinase Inhibitor. ChemMedChem 2018, 13, 2415-2426, 10.1002/cmdc.201800531.
- Linda Heintze; Dorian Schmidt; Theo Rodat; Lydia Witt; Julia Ewert; Malte Kriegs; Rainer Herges; Christian Peifer; Photoswitchable Azo- and Diazocine-Functionalized Derivatives of the VEGFR-2 Inhibitor Axitinib. International Journal of Molecular Sciences 2020, 21, 8961, 10.3390/ijms21238961.
- Wiktor Szymanski; Maria E. Ourailidou; Willem A. Velema; Dr. Frank J. Dekker; Dr. Ben L. Feringa; Light-Controlled Histone Deacetylase (HDAC) Inhibitors: Towards Photopharmacological Chemotherapy. Chemistry – A European Journal 2015, 21, 16517-16524, 10.1002/chem.201502809.
- Carlo Matera; Alexandre Gomila-Juaneda; Nuria Camarero; Michela Libergoli; Concepció Soler; Pau Gorostiza; Photoswitchable Antimetabolite for Targeted Photoactivated Chemotherapy. Journal of the American Chemical Society 2018, 140, 15764-15773, 10.1021/jacs.8b08249.
- Adrian Müller-Deku; Joyce C. M. Meiring; Kristina Loy; Yvonne Kraus; Constanze Heise; Rebekkah Bingham; Klara I. Jansen; Xiaoyi Qu; Francesca Bartolini; Lukas C. Kapitein; et al.Anna AkhmanovaJulia AhlfeldDirk TraunerOliver Thorn-Seshold Photoswitchable paclitaxel-based microtubule stabilisers allow optical control over the microtubule cytoskeleton. Nature Communications 2020, 11, 1-12, 10.1038/s41467-020-18389-6.
- Mickel J. Hansen; Willem A. Velema; Gerjan De Bruin; Herman S. Overkleeft; Wiktor Szymanski; Ben L. Feringa; Proteasome Inhibitors with Photocontrolled Activity. ChemBioChem 2014, 15, 2053-2057, 10.1002/cbic.201402237.
- Beatriz Blanco; Kathryn Palasis; Alaknanda Adwal; David F. Callen; Andrew D. Abell; Azobenzene-containing photoswitchable proteasome inhibitors with selective activity and cellular toxicity. Bioorganic & Medicinal Chemistry 2017, 25, 5050-5054, 10.1016/j.bmc.2017.06.011.
- Malgorzata Borowiak; Wallis Nahaboo; Martin Reynders; Katharina Nekolla; Pierre Jalinot; Jens Hasserodt; Markus Rehberg; Marie Delattre; Stefan Zahler; Angelika Vollmar; et al.Dirk TraunerOliver Thorn-Seshold Photoswitchable Inhibitors of Microtubule Dynamics Optically Control Mitosis and Cell Death. Cell 2015, 162, 403-411, 10.1016/j.cell.2015.06.049.
- Roberto Gaspari; Andrea E. Prota; Katja Bargsten; Andrea Cavalli; Michel O. Steinmetz; Structural Basis of cis - and trans -Combretastatin Binding to Tubulin. Chem 2017, 2, 102-113, 10.1016/j.chempr.2016.12.005.
- Ashton J. Engdahl; Edith A. Torres; Sarah E. Lock; Taylor Engdahl; Pamela S. Mertz; Craig N. Streu; Synthesis, Characterization, and Bioactivity of the Photoisomerizable Tubulin Polymerization Inhibitor azo-Combretastatin A4. Organic Letters 2015, 17, 4546-4549, 10.1021/acs.orglett.5b02262.
- Jonathon E. Sheldon; Martin Dcona; Charles E. Lyons; John Hackett; Matthew C. T. Hartman; Photoswitchable anticancer activity via trans–cis isomerization of a combretastatin A-4 analog. Organic & Biomolecular Chemistry 2015, 14, 40-49, 10.1039/c5ob02005k.
- Shiva K. Rastogi; Zhenze Zhao; Scott L. Barrett; Spencer D. Shelton; Martina Zafferani; Hailee E. Anderson; Madeleine O. Blumenthal; Lindsey R. Jones; Lei Wang; Xiaopeng Li; et al.Craig N. StreuLiqin DuWilliam J. Brittain Photoresponsive azo-combretastatin A-4 analogues. European Journal of Medicinal Chemistry 2018, 143, 1-7, 10.1016/j.ejmech.2017.11.012.
- Surya A. Reis; Balaram Ghosh; J. Adam Hendricks; D. Miklos Szantai-Kis; Lisa Törk; Kenneth N. Ross; Justin Lamb; Willis Read-Button; Baixue Zheng; Hongtao Wang; et al.Christopher SalthouseStephen J. HaggartyRalph Mazitschek Light-controlled modulation of gene expression by chemical optoepigenetic probes. Nature Chemical Biology 2016, 12, 317-323, 10.1038/nchembio.2042.
- Martin Reynders; Bryan S. Matsuura; Marleen Bérouti; Daniele Simoneschi; Antonio Marzio; Michele Pagano; Dirk Trauner; PHOTACs enable optical control of protein degradation. Science Advances 2020, 6, eaay5064, 10.1126/sciadv.aay5064.
- Patrick Pfaff; Kusal Samarasinghe; Craig M. Crews; Erick M. Carreira; Reversible Spatiotemporal Control of Induced Protein Degradation by Bistable PhotoPROTACs. ACS Central Science 2019, 5, 1682-1690, 10.1021/acscentsci.9b00713.
- Cyrille S. Kounde; Edward W. Tate; Photoactive Bifunctional Degraders: Precision Tools To Regulate Protein Stability. Journal of Medicinal Chemistry 2020, 63, 15483-15493, 10.1021/acs.jmedchem.0c01542.
- Dhanusha A. Nalawansha; Craig M. Crews; PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chemical Biology 2020, 27, 998-1014, 10.1016/j.chembiol.2020.07.020.
- Somiraa S. Said; Scott Campbell; Todd Hoare; Externally Addressable Smart Drug Delivery Vehicles: Current Technologies and Future Directions. Chemistry of Materials 2019, 31, 4971-4989, 10.1021/acs.chemmater.9b01798.
- W.F. Cheong; S.A. Prahl; A.J. Welch; A review of the optical properties of biological tissues. IEEE Journal of Quantum Electronics 1990, 26, 2166-2185, 10.1109/3.64354.
- Mingxin Dong; Amirhossein Babalhavaeji; Subhas Samanta; Andrew A. Beharry; G. Andrew Woolley; Red-Shifting Azobenzene Photoswitches for in Vivo Use. Accounts of Chemical Research 2015, 48, 2662-2670, 10.1021/acs.accounts.5b00270.
- Ron Siewertsen; -Ing. Hendrikje Neumann; Bengt Buchheim-Stehn; Rainer Herges; Christian Näther; Falk Renth; Friedrich Temps; Highly Efficient Reversible Z−E Photoisomerization of a Bridged Azobenzene with Visible Light through Resolved S1(nπ*) Absorption Bands. Journal of the American Chemical Society 2009, 131, 15594-15595, 10.1021/ja906547d.
- Miriam Schehr; Daniel Hugenbusch; Tobias Moje; Christian Näther; Rainer Herges; Synthesis of mono-functionalized S-diazocines via intramolecular Baeyer–Mills reactions. Beilstein Journal of Organic Chemistry 2018, 14, 2799-2804, 10.3762/bjoc.14.257.
- Nobuhiro Umeda; Hironori Takahashi; Mako Kamiya; Tasuku Ueno; Toru Komatsu; Takuya Terai; Kenjiro Hanaoka; Tetsuo Nagano; Yasuteru Urano; Boron Dipyrromethene As a Fluorescent Caging Group for Single-Photon Uncaging with Long-Wavelength Visible Light. ACS Chemical Biology 2014, 9, 2242-2246, 10.1021/cb500525p.
- Pratik P. Goswami; Aleem Syed; Christie L. Beck; Toshia R. Albright; Kaitlyn M. Mahoney; Ryan Unash; Emily Smith; Arthur H. Winter; BODIPY-Derived Photoremovable Protecting Groups Unmasked with Green Light. Journal of the American Chemical Society 2015, 137, 3783-3786, 10.1021/jacs.5b01297.
- Tomáš Slanina; Pradeep Shrestha; Eduardo Palao; Dnyaneshwar Kand; Julie A. Peterson; Andrew S. Dutton; Naama Rubinstein; Roy Weinstain; Arthur H. Winter; Petr Klán; et al. In Search of the Perfect Photocage: Structure–Reactivity Relationships in meso-Methyl BODIPY Photoremovable Protecting Groups. Journal of the American Chemical Society 2017, 139, 15168-15175, 10.1021/jacs.7b08532.
- Alexander P. Gorka; Roger R. Nani; Jianjian Zhu; Susan Mackem; Martin J. Schnermann; A Near-IR Uncaging Strategy Based on Cyanine Photochemistry. Journal of the American Chemical Society 2014, 136, 14153-14159, 10.1021/ja5065203.
- Julie A. Peterson; Chamari Wijesooriya; Elizabeth J. Gehrmann; Kaitlyn M. Mahoney; Pratik P. Goswami; Toshia R. Albright; Aleem Syed; Andrew S. Dutton; Emily A. Smith; Arthur H. Winter; et al. Family of BODIPY Photocages Cleaved by Single Photons of Visible/Near-Infrared Light. Journal of the American Chemical Society 2018, 140, 7343-7346, 10.1021/jacs.8b04040.
- Qiuning Lin; Lipeng Yang; Zhiqiang Wang; Yujie Hua; Dasheng Zhang; Bingkun Bao; Chunyan Bao; Xueqing Gong; Linyong Zhu; Coumarin Photocaging Groups Modified with an Electron-Rich Styryl Moiety at the 3-Position: Long-Wavelength Excitation, Rapid Photolysis, and Photobleaching. Angewandte Chemie International Edition 2018, 57, 3722-3726, 10.1002/anie.201800713.
- D. Warther; S. Gug; Alexandre Specht; F. Bolze; J.-F. Nicoud; Alexandre Mourot; M. Goeldner; Two-photon uncaging: New prospects in neuroscience and cellular biology. Bioorganic & Medicinal Chemistry 2010, 18, 7753-7758, 10.1016/j.bmc.2010.04.084.
- Chongzhao Ran; Zhaoda Zhang; Jacob Hooker; Anna V Moore; In Vivo Photoactivation Without “Light”: Use of Cherenkov Radiation to Overcome the Penetration Limit of Light. Molecular Imaging and Biology 2011, 14, 156-162, 10.1007/s11307-011-0489-z.
- Ivan Tochitsky; Jay Trautman; Nicholas Gallerani; Jonatan G. Malis; Richard H. Kramer; Restoring visual function to the blind retina with a potent, safe and long-lasting photoswitch. Scientific Reports 2017, 7, srep45487, 10.1038/srep45487.
- F. Reeßing; M. C. A. Stuart; D. F. Samplonius; R. A. J. O. Dierckx; B. L. Feringa; W. Helfrich; W. Szymanski; A light-responsive liposomal agent for MRI contrast enhancement and monitoring of cargo delivery. Chemical Communications 2019, 55, 10784-10787, 10.1039/c9cc05516a.
- Sneha S. Kelkar; Theresa M. Reineke; Theranostics: Combining Imaging and Therapy. Bioconjugate Chemistry 2011, 22, 1879-1903, 10.1021/bc200151q.
- Twan Lammers; Silvio Aime; Wim E. Hennink; Gert Storm; Fabian Kiessling; Theranostic Nanomedicine. Accounts of Chemical Research 2011, 44, 1029-1038, 10.1021/ar200019c.
- Oleg Babii; Sergii Afonin; Tim Schober; Liudmyla V Garmanchuk; Liudmyla I Ostapchenko; Volodymyr Yurchenko; Sergey Zozulya; Oleksandr Tarasov; Iryna Pishel; Anne S Ulrich; et al.Igor V Komarov Peptide drugs for photopharmacology: how much of a safety advantage can be gained by photocontrol?. Future Drug Discovery 2020, 2, FDD28, 10.4155/fdd-2019-0033.

