 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Orly Weissberg | + 8059 word(s) | 8059 | 2021-08-03 05:31:06 | | | |
| 2 | Orly Weissberg | + 12 word(s) | 8071 | 2021-08-19 12:35:59 | | | | |
| 3 | Vivi Li | -5 word(s) | 8066 | 2021-08-20 10:14:05 | | |
Video Upload Options
Chromodomain-helicase-DNA-binding protein 8 (CHD8) has been identified as one of the genes with the strongest association with autism. The CHD8 protein is a transcriptional regulator that is expressed in nearly all cell types and has been implicated in multiple cellular processes, including cell cycle, cell adhesion, neuronal development, myelination, and synaptogenesis. Considering the central role of CHD8 in the genetics of autism, a deeper understanding of the physiological functions of CHD8 is important to understand the development of the autism phenotype and potential therapeutic targets. Different CHD8 mutant mouse models were developed to determine autism-like phenotypes and to fully understand their mechanisms.
1. Introduction to Autism
Autism spectrum disorder (ASD) is a heterogeneous neurodevelopmental disorder characterized by impaired sociability and language development, and repetitive and stereotypic behaviors. Autism has a strong genetic etiology, including involvement of chromatin rearrangements, de novo mutations, and common variants [1][2][3]. An interaction of multiple genetic factors and environmental factors may be involved in the development of ASD [4]. ASD incidence is steadily rising in the world population [5]. The prevalence is estimated to be one in 54 children at 8 years of age by the Centers for Disease Control and Prevention (CDC), or one in 40 at ages 3 to 17 years of age according to the National Survey of Children’s Health (NSCH) and the National Health Interview Survey. There is a male bias in the diagnosis of autism in the order of 4.3:1 in the USA [6]. The cost of treating ASD individuals including educational support, loss of parent working days, special health services, and others was USD 268 billion in 2015, and estimated to be USD 461 billion in 2025 in the USA [6][7][8]. The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) defines autism using social communication impairment, restricted interests, and repetitive behaviors. Comorbidities include anxiety, epilepsy, attention deficit hyperactivity disorder (ADHD), speech and language delay, sleep disorders, gastrointestinal problems, and impaired learning and motor difficulties [9][10][11][12][13][14]. To date, there is no biological diagnostic assay [15][16][17] or approved curative treatment [18][19]. ASD etiology is poorly understood [20], and it is now accepted that it includes different subtypes induced by different etiologies and pathways, including genetic and environmental factors [12].
2. Association of CHD8 and Autism
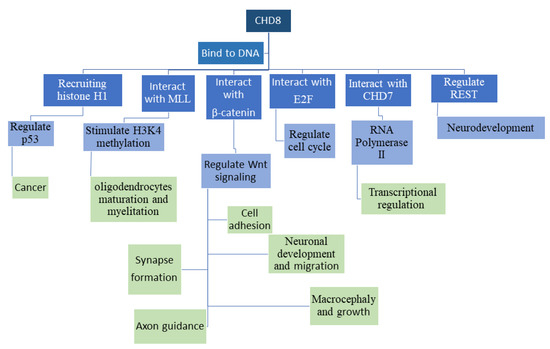
3. Basic Molecular Functions of CHD8
4. Expression of CHD8 and Knockdown of CHD8 in In Vitro or Nonmammalian Model Systems
5. Effects of CHD8 on Non-Neuronal Tissues
Several other studies have shown a role for CHD8 regulation in non-neuronal cells in both behavioral and physiological pathways. Slenderness is one of symptoms seen in individuals with CHD8 mutation. Kita et al. determined that CHD8 deletion specifically in preadipocytes leads to decrease in adipogenesis and a slender phenotype in mice. They found that CHD8 interaction with C/EBPα and PPARɤ is crucial for adipogenesis and increase of white adipose tissue mass. Therefore, CHD8 mutations may lead to physiological changes through regulation in peripheral tissues [59]. Separate studies found that oligodendrocyte precursor cell (OPC) proliferation, differentiation, and survival is regulated by CHD8 and CHD7. CHD8 interacts in OPCs with ASD risk factor genes, therefore it is suggested that oligodendrocytes take part in ASD pathology [60]. In oligodendrocytes from Chd8 OPC knockout mice (Chd8flox/flox; Olig1-Cre+), mutants displayed dysregulation in OPC differentiation, proliferation, and myelination. By recruiting KMT2 histone methyltransferase and H3K4, CHD8 regulates the BRG1-CHD7 cascade (BRG1activate CHD7) that is responsible for oligodendrocyte development. In addition, these mice die at P21, which is the peak time of myelination. Hence, it suggested that CHD8 is crucial for myelination regulation and OPC differentiation. In addition, GO analysis revealed CHD8 influences neurogenesis, gliogenesis, Wnt signaling, and apoptosis [45]. CHD8 inhibition resulted in macrocephaly [48] due to increase of forebrain and midbrain and impairment of postmitotic enteric neuron results in gastrointestinal dysfunction in zebrafish [23].
6. Mouse Models in the Research of CHD8, Neurodevelopment, and Autism
7. Interplay between CHD8 and Other Genetic or Environmental Factors
8. Future
CHD8 is expressed in various cell types; however, it has cell-type-specific functions, as seen in the models described above [71]. It is expressed in higher levels during the embryonic and postnatal stages, hence it is important for development [51]. Many studies had emphasized CHD8 involvement in ASD in a way that it might be considered a separate ASD subtype. However, multiple questions remain open and need further study. The differential behavioral phenotypes seen in the models discussed above further suggest that different mutations may have separate effects on CHD8 function. The different phenotypes may be partly due to laboratory conditions, but are also likely due to disruption of different areas of the CHD8 gene. In addition, it is not clear if the role of CHD8 in neuronal activity is completely developmental, or if there are specific roles for CHD8 in brain function past development. In order to answer these questions, further study is needed.[72]
|
Table 1: known CHD8 mutations. |
|||||||||
|
AA change |
S.M |
Chr14 P |
NVIQ |
VIQ |
FSIQ |
INT |
Mut |
Phen |
|
|
O’Roak et al. 2012 (1) |
p.Gln959TERM |
34 |
DN |
n/a |
ASD |
||||
|
Brian J. O’Roak et al. 2012 (2) |
p.Ser62X |
c.185C>G |
21899618 |
78 |
DN |
Ns |
ASD |
||
|
p.Tyr747X |
21878133 |
38 |
DN |
Fs |
ASD+ID |
||||
|
c.3519-2A>G |
47 |
37 |
43 |
DN |
Sp |
ASD |
|||
|
p.Gln1238X |
c.3712C>T |
21871178 |
34 |
75 |
74 |
DN |
Ns |
ASD+ID |
|
|
p.Arg1337X |
c.4009C > T |
21870169 |
92 |
DN |
Ns |
ASD |
|||
|
p.Glu2103ArgfsX3 |
c.6307_6310del |
21861643 |
67 |
DN |
Fs |
ASD |
|||
|
p.Leu2120ProfsX13 |
c.6359_6360del |
21861376 |
93 |
DN |
Fs |
ASD |
|||
|
p.Asn2371LysfsX2 |
c.7112_7113insA |
21859175 |
19 |
DN |
Fs |
ASD+ID |
|||
|
p.His2498del |
98 |
DN |
Aa |
ASD |
|||||
|
Bernier et al. 2014 (3) |
p.Val984X |
IN |
Fs |
ASD/ID |
|||||
|
p.Glu1114X |
c.3340G>T |
41 |
27 |
34 |
DN |
Ns |
ASD |
||
|
p.Glu1932SerfsX3 |
DN |
Fs |
ASD/ID/DD |
||||||
|
p.Glu2136ArgfsX6 |
<40 |
DN |
Fs |
ID |
|||||
|
p.Lys2287 del |
n/a |
Aa |
ID/ADHD |
||||||
|
p.Arg910Gln |
c.2729G>A |
27 |
DN |
Mns |
ASD+ID |
||||
|
p.Gly1710Val |
c.5129G>T |
IN |
Mns |
ASD+DD |
|||||
|
p.Arg1797Gln |
c.5390G>A |
IN |
Mns |
ASD |
|||||
|
Prontera et al. 2014 (4) |
~114Kb microdeletion |
21,823,852 –21,937,621 |
76 |
DN |
Md |
ASD |
|||
|
B. J. O’Roak et al. 2014 (5) (Supplementary) |
p.Arg212Gln |
72 |
DN |
Ms |
ASD |
||||
|
p.Gln696Lys |
125 |
88 |
DN |
Ms |
|||||
|
p.Met904Ile |
63 |
DN |
Ms |
ASD+ID |
|||||
|
p.Arg1834X |
c.5500C>T |
93 |
DN |
Ns |
ASD+ID |
||||
|
p.Arg1580Trp |
74 |
97 |
DN |
Ms |
ASD |
||||
|
c.4818-2A>C |
103 |
96 |
DN |
Ns |
ASD |
||||
|
c.5051 + 2T>A |
DN |
Ns |
ASD |
||||||
|
Iossifov et al. 2014 (6) |
c.1593_1601_38del |
DN |
Ssv |
ASD |
|||||
|
De Rubeis et al. 2014 (7) |
p.Leu834Pro |
DN |
Ms |
ASD |
|||||
|
p.Arg1242Gln |
DN |
Ms |
ASD |
||||||
|
p.Gly1602ValfsX15 |
DN |
Ssv-Fs |
ASD |
||||||
|
p.Ser1606ArgfsX8 |
DN |
Ssv-Fs |
ASD |
||||||
|
p.Tyr1642LeufsX25 |
DN |
Ssv-Fs |
ASD |
||||||
|
Talkowski et al. 2012 (8) |
t.14q11.2; 3q25.31 |
DN |
Tr |
ASD+ID |
|||||
|
McCarthy et al. 2014 (9) |
p.Ser2173X |
c.6518C>A |
21860919 |
DN |
Ns |
ASD+SHZ |
|||
|
Kimura et al. 2016 (10) |
p.His1439del |
50 |
DN |
SCZ |
|||||
|
p.Lys2287del |
DN |
SCZ |
|||||||
|
p.Arg2333Cys |
50 |
DN |
ASD+ID+DD+SCZ |
||||||
|
p.Arg773Gln |
DN |
ASD+SCZ |
|||||||
|
Merner et al. 2016 (11) |
p.Asn2092LysfsX2 |
c.6276dup |
21859176 |
56 |
DN |
Fs |
ASD+ID |
||
|
p.Arg7Cys |
c.19C>T |
21897482 |
IN |
Pms |
ASD |
||||
|
p.Ile1325Thr |
c.3974T>C |
21868146 |
IN |
nSn |
SCZ |
||||
|
p.Glu1750Lys |
5248G>A |
21861869 |
n/a |
nSn |
ASD |
||||
|
p.Arg1879Cys |
c.5635C>T |
21860965 |
IN |
nSn |
SCZ |
||||
|
p.Arg1901Cys |
c.5701C>T |
21860899 |
n/a |
nSn |
SCZ |
||||
|
p.Gly1998Ala |
c.5993G>C |
21860047 |
n/a |
nSn |
ID |
||||
|
p.Arg2035Gln |
c.6104G>A |
21859746 |
IN |
nSn |
SCZ |
||||
|
p.Gly162Gly |
G/C |
21896306 |
IN |
Sn |
ASD |
||||
|
p.Leu1305Leu |
A/G |
21868205 |
IN |
Sn |
SCZ |
||||
|
p.Ala1693Ala |
A/G |
21862038 |
IN |
Sn |
SCZ |
||||
|
p.Glu1825Glu |
G/A |
21861642 |
IN |
Sn |
ASD |
||||
|
p.His1989His |
C/T |
21860073 |
IN |
Sn |
ID |
||||
|
p.Asp2261Asp |
C/T |
21853898 |
IN |
Sn |
ASD |
||||
|
Smyk et al. 2016 (12) |
~445 kb microduplication |
21,507,092-21,952,439 |
DN |
Md |
DD+HDAD |
||||
|
Zahir et al. 2007 (13) |
~101 kb microduplication |
20,896,740 – 20,998,178 |
DN |
Md |
DD+ID |
||||
|
~1.6 Mb microduplication |
19,584,863 – 21,207,935 |
DN |
Md |
DD+ID |
|||||
|
~1.079 Mb microduplication |
19,853,310 – 20,932,827 |
DN |
Md |
DD+ID |
|||||
|
Terrone et al. 2014 (14) |
~2.89 Mb microduplication |
chr14: 19,788,445 - 22,675,219 |
DN |
Md |
ASD |
||||
|
T. Wang et al. 2016 (15) |
p.Asp691GIy |
DN |
Ms |
ASD |
|||||
|
p.Lys750AsnfsX14 |
DN |
LGD |
ASD |
||||||
|
p.Asn1235MetfsX18 |
DN |
LGD |
ASD |
||||||
|
p.Arg1897ThrfsX23 |
21862265 |
DN |
LGD |
ASD |
|||||
|
Stolerman et al. 2016 (16) |
Exons 26-28 deletion |
21,863,796-21,868,103 |
DN |
ASD+DD+ID |
|||||
|
Han et al. 2018 (17) |
p.Arg1551Cys |
c.4651C>T |
65 |
DN |
Ms |
ID |
|||
|
J. Wang et al. 2018 (18) |
p.Glu883X |
c.2647C > A |
DN |
Ns |
ASD+DD |
||||
|
p.Met559Ile |
c.1677C > A |
DN |
Ms |
ASD+DD |
|||||
|
D’Gama et al. 2015 (19) |
p.Val744Ile |
c.2230G > A |
DN |
Ms |
ASD+ID |
||||
|
Cappi et al. 2016 (20) |
p.Elu1327Lys |
C > T |
21870199 |
DN |
Ms |
ID+OCD |
|||
|
Arnett et al. 2018 (21) |
p.Glu1727X |
c.5179G > T |
DN |
Sg |
ASD |
||||
|
p.Arg1402X |
c.4204C > T |
DN |
Sg |
ASD |
|||||
|
p.Ile1108AsnfsX7 |
c.3322_3323insA |
DN |
Fs |
ASD |
|||||
|
p.Asn807ThrfsX78 |
c.2420del |
DN |
Fs |
ASD |
|||||
|
p.Arg952X |
c.2854C > T |
DN |
Sg |
ASD |
|||||
|
p.His782ProfsX7 |
c.2345del |
DN |
Fs |
ASD |
|||||
|
c.3882 + 1G > A |
DN |
Sp |
ASD |
||||||
|
Wong et al. 2019 (22) |
p.Pro165Leu |
c.494C>T |
DN |
Ms |
ASD |
||||
|
Yasin et al. 2019 (23) |
~33.269 kb deletion |
21,827,942-21,861,211 |
DN |
ASD+ID+DD |
|||||
|
Ostrowski et al. 2019 (24) |
c.470del |
DN |
Fs |
ID |
|||||
|
c.517_533del |
DN |
Fs |
ID |
||||||
|
p.Arg564X |
c.1690c>T |
DN |
Sg |
ASD+ID |
|||||
|
c.1899+1G>T |
DN |
Ss |
ASD+ID |
||||||
|
c.2024+5G>A |
DN |
Sg |
ID |
||||||
|
p.Glu714X |
c.2140G>T |
DN |
Sg |
ASD+ID |
|||||
|
p.Tyr854X |
c.2562_2563del_p. |
DN |
Sg |
ASD+ID |
|||||
|
p.Asn873Asp |
c.2617A>G |
DN |
Sg |
ASD+ID |
|||||
|
p.Thr976Lys |
c.2927C>A |
DN |
Fs |
ID |
|||||
|
c.3011_3012del |
DN |
Sp |
ASD+ID |
||||||
|
c.3518+5G>C |
DN |
Fs |
ASD+ID |
||||||
|
c.3528_3529insAA |
Inherited |
Fs |
ID |
||||||
|
c.3569_3587del |
DN |
Sg |
ID |
||||||
|
c.4093_4094del |
DN |
Fs |
ASD+ID |
||||||
|
p.Arg1472X |
c.4414C>T |
DN |
Sg |
ASD+ID |
|||||
|
p.Ser1420X |
c.4259_4260del |
DN |
Fs |
ID |
|||||
|
c.5386del |
IN |
Sg |
ID |
||||||
|
c.5599+2T>C |
N/A |
Fs |
ID |
||||||
|
c.6115del |
n/a |
Fs |
ID |
||||||
|
c.7511dup |
DN |
Fs |
ID |
||||||
|
Cotney et al. 2015 (18,25) |
p.Arg286Cys |
c.856C > T |
Missense |
ASD |
|||||
|
p.Arg2158Cys |
c.6472C > T |
Missense |
ASD |
||||||
|
p.Arg2180Cys |
c.6538C > T |
Missense |
ASD |
||||||
|
p.G2277Ala |
c.6830G >C |
Missense |
ASD |
||||||
|
p.Arg2314Gln |
c.6941G > A |
Ns |
ASD |
||||||
|
Siu et al. 2019 (26) |
p.Arg1173Gly |
c.3517 A>G |
DN |
ASD |
|||||
|
p.Asn740Ser |
c.2219A>G |
IN |
ASD |
||||||
|
p.Pro2281Ala |
c.6841C>G |
IN |
ASD |
||||||
|
p.Gly2189Arg |
c.6565G>A |
IN |
ASD |
||||||
|
p.Ala1314Thr |
c.3940G>A; |
IN |
ASD |
||||||
|
p.His2500Pro |
c.7499A>C |
n/a |
ASD |
||||||
|
p.Thr2050fs |
c.6148dupA |
DN |
ASD |
||||||
|
c.4215G>T |
ID |
Sn |
ASD |
||||||
|
p.Arg2217 |
c.6649C>T |
ID |
ASD |
||||||
|
p.Pro2316LeufsX39 |
c.6947delC |
n/a |
ASD |
||||||
|
p.Arg1443Cys |
c.4327C>T |
ID |
ASD |
||||||
|
Douzgou et al. 2019 (27) |
p.Ser1420X |
c.4259_4261GTC>G |
21869143-2186914 |
n/a |
Sg |
ASD+ID |
|||
|
p.Glu1004ValfsX22 |
c.3011_3012GA>T |
21873918-21873920 |
DN |
Fs |
DD |
||||
|
c.1899+1G |
21883883-21883883 |
DN |
Sp |
ASD+ID |
|||||
|
p.Lys545AsnfsX47 |
c.1635delCA>C |
21894367-21894368 |
n/a |
Fs |
ASD+ID |
||||
|
p.Arg1242X |
c.3724C>T_ |
21870653-21870653 |
DN |
Sg |
ASD+ID+DD |
||||
|
p.Gln687X |
c.2059C>T |
21882543-2188254 |
DN |
Sg |
ASD+ID |
||||
|
p.Tyr902X |
c.2706GT>G |
21876494-2187649 |
DN |
Sg |
ASD |
||||
|
p.Tyr854X |
c.2562_2563del |
21876637-21876639 |
DN |
Sg |
ASD+ID |
||||
|
c.3518+1 |
21877595 -21877595 |
DN |
Sp |
ID |
|||||
|
p.Leu1206X |
c.3617T>G |
21871273- -21871273 |
DN |
Sg |
ASD+ID |
||||
|
Smol et al. 2020 (28) |
401 kb microduplication |
21499240 -21899985 × 3 |
Md |
ASD+DD+ID |
|||||
|
277 kb duplication |
21622823 -21899759 × 3 |
DD |
|||||||
|
Tran et al. 2020 (29) |
p.Ile1192Thr |
c.3575 T > C |
21871315 |
DN |
Ms |
ASD |
|||
|
Wu et al. 2020 (30) |
p.G1602Vfs*13 |
c.4800delA |
21399998- 21399998 |
79 |
DN |
Fs |
ASD |
||
|
p.N885Tfs*14 |
c.2654delA |
21408388- 21408388 |
IN |
Fs |
ASD |
||||
|
Alotaibi and Ramzan 2020 (31) |
p.Arg1662Ter |
c.4984C>T |
DN |
Pms |
ASD+DD+ID |
||||
|
An et al. 2020 (32) |
p.Tyr1168Asn |
c.3502 T>A |
DN |
Ms |
ASD |
||||
|
p.Arg1188X |
c.3562C>T |
DN |
Ns |
ASD+ID |
|||||
|
c.4818-1G>A |
DN |
Sp |
ASD |
||||||
|
p.Glu689X |
c.2065C>A |
DN |
Ns |
ASD+DD |
|||||
|
p.Lys2286Arg |
c.6857A>G |
Ms |
ASD |
||||||
|
p.Arg773Gln |
c.2318G>A |
IN |
ASD |
||||||
|
p.Val2521Ala |
c.7562T>C |
IN |
ASD |
||||||
Abbreviations: AA change- amino acid change, S.M-sequencing mutation, Chr14 P- chromosome 14 position, NVIQ- non-verbal I.Q, VIQ- verbal I.Q, FSIQ- Full Scale I.Q, INT- inherited type, MutT-mutation type, Phen- phenotype. DN- de novo, IN- inherited, Fs-frameshifting indel, Ns-nonsense, Sp-splice-site, Aa- single amino acid deletion, Ms-missense, Mns- Missense near splice site, Tr- Translocation, Ssv- Splice site variant, nSn- non-synonymous, Sn- synonymous, Sg- stop-gained, Pms- premature stop codon, Md- microdeletion, Dup- duplication, LGD- likely gene-disruptive, ASD- autism spectrum disorder, ID- intellectual disability, DD- developmental delay, SCZ- Schizophrenia. Fs-frameshift, X-stop, X23-stop after 23 amino acid, Ter- stop, Arg 1023del- amino acid Arg in position 1023 was deleted.
|
Table 2: CHD8 mouse models. |
||||||||
|
Mouse model |
Chd8+/ΔSL and Chd8+/ΔL |
Chd8+/- |
Chd8+/del5 |
Chd8+/− |
Chd8+/N2373K |
Chd8V986*/+ |
Chd8+/E31T |
Olig1-Cre/Chd8F/F mice |
|
Reference |
Katayama et al. 2016 (1) |
Platt et al. 2017 (2) |
Gompers et al. 2017 (3) |
Suetterlin et al. 2018 (4) |
Jung et al. 2018 (5) |
Jiménez et al. 2020 (7) |
Hulbert et al. 2020 (8) |
Kawamura et al. 2020 (9) |
|
Mutation |
Deletion of Exons 12–14 (“ΔL”) And Deletion of Exons 2–10 (“ΔSL”) |
A 7-nucleotide deletion in exon 1 that causes a frameshift mutation leading to loss-of-function |
Novel germline 5 bp deletion in Chd8 exon 5. |
Early frameshift and termination of translation at amino acid 419 at exon 3. Chd8flox/+ crossed with β-actinCre mice to generate β-actinCre;Chd8+/− mice. |
Asn2373LysfsX2 in mice parallel to Asn2371LysfsX2 in humans. In exon 37. |
Stop codon at the valine 986 which equivalent to valine 984 in mouse Chd8. |
Gene trap inserted after Exon 31. |
Deletion of Exons 12–14 (“ΔL”) in oligodendrocytes |
|
Brain volume |
Increased (at E18.5 and adult) |
n/a |
Increased Maximal cortical anteroposterior length of Chd8+/del5 brains was ~7% longer at P0 (no substantial differences between sexes), 7.5% increase in absolute volume of cortex, whole-mount and Nissl- stained coronal brain sections at P7- no neuropathological anomalies were observed, cortical thickness at 30% and 70% distance from the dorsal midline- no significant differences. The overall neocortical section area was ~8% larger, cerebral white matter and cerebral gray matter were larger at 5.4% and 6.1% respectively. Robust increases in absolute volume across cortical regions, hippocampus (+10.3%) and amygdala (+11.0%). Increased cortical thickness. Deep cerebellar nuclei showed decreased relative volume (-1 to -3%). |
Increased Total brain increased by 2.7%, several brain regions, including cortical areas, hippocampus and parts of the cerebellum showed volumetric increases |
Increase (MRI) |
Increased (P0) |
n/a |
No differ |
|
Brain morphology and function (MRI, μCT, rsfMRI) |
n/a |
Morphological analysis using Nissl staining shows no overt phenotype present in the somatosensory cortex no increase in the number of cortical progenitor cells as measured by BrdU incorporation within the somatosensory cortex no increase in either the total cell-cycle length or the length of S phase within the somatosensory cortex (no gross de- fects in specification, migration, or lamination of different sub- types in the neocortex.) increase in both intraocular distance as well as total brain volume (10W) (MRI) |
Increased absolute volume of cerebral cortex (f-stat = 33.6, FDR < 0.1%), hippocampus (f-stat = 29.0, FDR < 0.1%) and amygdala (f-stat = 38.6, FDR < 0.1%) increased brain volume (MRI) |
interorbital distance- significantly wider anterior–posterior length of the interparietal bone – increased, suggestive of more wide-spread craniofacial anomalies (μCT) hotspots for increased connectivity in Cortical and Hippocampal Networks (entorhinal, retrosplenial, auditory cortical and posterior hippocampal areas), increased cortical connectivity in auditory regions, increase in connectivity between ventral hippocampus and auditory cortical regions, increased connectivity of this region with both cingu- late and entorhinal cortices (rsfMRI) |
anterior cingulate, anterior commissure, and cerebellum in female (not in male) Gross brain morphology- normal hippocampus displays sexually dimor- phic synaptic transmission and neuronal firing (MRI) |
n/a |
length increase for adult mice |
number of oligodendrocytes was significantly reduced in the corpus callosum of Olig1-Cre/Chd8L F/F mice at P7 and P14 |
|
Body weight |
No differ |
Decrease (10W) |
No differ |
Decrease (5W) |
No differ |
No differ at P0 Decrease at 25W |
n/a |
No differ |
|
Transcriptomic changes in the brain |
||||||||
|
* RNA-seq |
n/a |
Brain development, epigenome regulation, neuronal and synaptic adhesion. |
Forebrain at E12.5, E14.5, E17.5, P0 and adult mice Chd8 expression declined across development. Decreased in genes of RNA processing, chromatin remodeling, and cell cycle, increased of genes linked to immune function and cell identity of astrocytes or microglia. Alteration of genes involved in neuronal maturation Increased prenatal proliferation of neural progenitors, |
Neocortical tissue at E12.5 and P5 Upregulated - KEGG pathways related to protein transport, the ribosome and oxidative phosphorylation downregulated genes related to cell adhesion, axonal guidance and calcium signaling pathways, Suz12 targets genes. At P5 significant enrichment of cell adhesion and axonal guidance genes in the downregulated genes. axon guidance and cell adhesion genes that are preferentially expressed in CA2 and auditory areas at adult stages and whose expression is dysregulated at P5 |
Hippocampus at P0, P25 Chd8 expression was stronger than at later stages No differentially expressed genes (DEGs) in P0 in males or females. Sexually dimorphic enrichment patterns at P25, there were three DEGs in males and 96 DEGs in females. In P25 females- extracellular vesicles, including blood microparticles and extracellular exosomes, platelet activation. P25 female whole-brain enriched genes were ‘blood microparticle’ and ‘platelet’. P25 male, but not female, whole-brain were strongly and negatively enriched genes for ‘synapse’. Males and females showed largely similar enrichment patterns for ‘ribosome’ and ‘mitochondria/oxidoreduc- tase’ genes |
Cerebral cortex at E14.5, 1M, 6M, 12M Chd8 expression was highest at E14.5 and persisted at a lower level throughout life genes associated with focal adhesion, neurodevelopmental, proteostasis, sodium channel activity and synaptic function were reduced reduced mTORC1 and IRE1 pathway activation decreased XBP1 expression genes associated with heat shock factor 1 (HSF1) signaling and chaperone function were reduced c- MET signaling pathways were upregulated protein homeostasis is impaired |
n/a |
oligodendrocyte-specific genes were down-regulated |
|
* ChIP-seq |
Expression of genes related to synapses and ion was downregulated in Chd8+/ΔL mice. Neural development is delayed during the early to mid-fetal stage in the mutant mice |
CHD8 binding sites are enriched in promoters. Enrichment for histone and chromatin modification as well as al- terations in mRNAand protein processing. |
Strong concordance in enriched functional annotation terms between DE and Chd8-bound genes. Strong enrichment for binding among downregulated genes. |
n/a |
n/a |
n/a |
n/a |
significant enrichment for myelination, membrane, lipid metabolic process and sterol biosynthesis process. |
|
Repetitive behavior |
||||||||
|
*Self-grooming |
No differ |
No differ |
No differ |
No differ |
Increase in adult male when isolated for 3 d, but showed normal self-grooming and other repetitive behaviors when housed together, whereas females showed no isolation-induced self-grooming |
n/a |
No differ |
|
|
* Marble burying |
n/a |
No differ |
No differ |
No differ |
No differ |
No differ |
n/a |
|
|
Anxiety like behavior |
Increased |
Increased |
n/a |
In adult male- no differ |
n/a |
No differ |
Increased |
|
|
Learning impairments |
||||||||
|
* Light–dark emergence task |
Decrease |
Increased in the latency to enter the light side |
n/a |
No differ |
n/a |
n/a |
No differ |
n/a |
|
* Elevated plus maze |
Decrease |
n/a |
n/a |
n/a |
No differ |
No differ (6M) |
No differ |
No differ |
|
* Elevated zero maze |
n/a |
n/a |
n/a |
n/a |
n/a |
n/a |
No differ |
n/a |
|
* T-maze forced-alternation test |
No differ Chd8+/ΔL mice |
n/a |
n/a |
n/a |
n/a |
n/a |
n/a |
n/a |
|
* T-maze left–right discrimination test |
percentage of correct responses was reduced for Chd8+/ΔL mice |
n/a |
n/a |
n/a |
n/a |
n/a |
n/a |
n/a |
|
* Open field |
Center time – Decrease Total distance- no differ |
Center time – Decrease Total distance- Decrease Reduced locomotion |
No differ |
Hypoactivity No evidence of anxiety Total distance- Decrease Center time- no differ |
Hypoactivity and decreased center time in adult male but not female Total distance- decrease Center time- decrease |
Distance traveled. No significant differences Number of rears - significant decrease Time in center. No significant differences at 6M but significant decrease in center time at 12M |
No differ |
No differ |
|
* Acoustic startle test |
Amplitude- Decrease Prepulse inhibition- Increase |
n/a |
n/a |
n/a |
Prepulse inhibition- no differ |
Amplitude - no differ Prepulse inhibition- no differ |
n/a |
No differ |
|
*Contextual fear conditioning |
n/a |
No differ |
Deficits in learning and memory, less freezing, lower freezing scores to the auditory cue |
n/a |
n/a |
n/a |
n/a |
n/a |
|
* Tone fear conditioning |
n/a |
No differ |
n/a |
n/a |
n/a |
n/a |
n/a |
n/a |
|
* Novel object recognition |
Decrease |
n/a |
Deficits in recognition |
n/a |
n/a |
n/a |
n/a |
n/a |
|
*Morris water maze test (Spatial learning abilities and cognitive flexibilit) |
n/a |
n/a |
n/a |
Normal in the learning part, normal cognitive, spatial learning abilities and flexibility. |
n/a |
n/a |
n/a |
n/a |
|
* T-maze left-right discrimination test |
n/a |
n/a |
n/a |
n/a |
n/a |
n/a |
n/a |
No differ |
|
Olfactory |
n/a |
n/a |
n/a |
Increased interest in an odour with social significance |
in adult male- no differ |
No differ |
n/a |
No differ |
|
Motor function |
||||||||
|
* Rotarod |
increase in acquired motor learning |
n/a |
n/a |
normal motor abilities |
in adult male- no differ |
n/a |
increase in motor performance |
n/a |
|
*Forelimb grip strength |
n/a |
n/a |
n/a |
slightly but significantly reduced |
n/a |
n/a |
n/a |
n/a |
|
Sociability |
||||||||
|
Social communication |
n/a |
Mild deficit in social interaction behavior in the social novelty but not the sociability test of the three- chambered social approach task |
No differ |
No obvious communication deficit |
n/a |
Increased social preference |
No differ |
Mild deficit in social-novelty preference but not in sociability |
|
*Three-chamber test for social novelty preference |
Sociability- no differ Novelty preference- Decrease |
Sociability and Entries- no differ Novelty preference- Decrease |
No differ |
Normal sociability minor deficits in social novelty |
In adult male- no differ Sociability and Novelty preference- no differ |
Sociability- no duffer Novelty preference – increased a shift in preference to the newly introduced stranger (6M+12M) Entries-no differ |
No differ |
Significant preference for a novel mouse |
|
*social-interaction test |
reduced for mutant mice duration per contact was greatly increased |
n/a |
n/a |
n/a |
in adult male- no differ (dyadic social interaction) |
n/a |
n/a |
Social contacts did not differ duration per contact and total contact time-increased |
|
* Communication through USVs ultrasonic vocalizations (USVs) |
n/a |
n/a |
No differ |
No differ |
More frequently, rapidly, and for longer durations in male P5-11 but not in female (when separated from their mothers) In adult male- no differ |
Increased social interest |
No difference in the number of calls made, increased call duration |
n/a |
|
Cognitive impairments |
||||||||
|
* Operant conditioning task |
n/a |
n/a |
n/a |
n/a |
n/a |
n/a |
No differ |
n/a |
|
Maternal-homing test |
n/a |
n/a |
n/a |
n/a |
Spent more time with the reunited mothers, suggesting enhanced mother-attachment behavior in male but not female |
n/a |
n/a |
n/a |
|
Homozygous embryonic lethal |
+ |
+ |
+ |
n/a |
n/a |
+ |
+ |
Normal at birth, although died before 3 weeks |
|
Other |
*Shorter intestine and tended to manifest slower intestinal transit |
*Using CRISPR/Cas9 *Synaptic Dysfunction within MSNs in the NAc *local decrease of inhibitory transmission may contribute to the enhanced excitatory inputs onto MSNs in the NAc *CHD8 expression in adults is not required for the increased anxiety-like or decreased locomotor behavior but is required for acquired motor learning in the ro- tarod performance test. |
*Using CRISPR/Cas9 |
n/a |
*In adult male- no differ- nest building and sleeping (huddling). *Opposite changes in inhibitory synaptic transmission in the male and female Chd8+/N2373K Hippocampus |
*Using CRISPR/Cas9 * Pup survival at P2- reduced when litters were reared by Chd8V986*/+ dams |
*Defective myelin formation *Nest-building test and Porsolt forced-swim test - no differ |
|
|
Reference |
Katayama et al. 2016 (1) |
Platt et al. 2017 (2) |
Gompers et al. 2017 (3) |
Suetterlin et al. 2018 (4) |
Jung et al. 2018 (5) |
Jiménez et al. 2020 (7) |
Hulbert et al. 2020 (8) |
Kawamura et al. 2020 (9) |
References
- Grønborg, T.K.; Schendel, D.E.; Parner, E.T. Recurrence of autism spectrum disorders in full- and half-siblings and trends over time: A population-based cohort study. JAMA Pediatr. 2013, 167, 947–953.
- American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders: DSM-5, 5th ed.; The American Psychiatric Association: Washington, DC, USA, 2014; Volume 11, ISBN 01679236 (ISSN).
- Krumm, N.; Turner, T.N.; Baker, C.; Vives, L.; Mohajeri, K.; Witherspoon, K.; Raja, A.; Coe, B.P.; Stessman, H.A.; He, Z.X.; et al. Excess of rare, inherited truncating mutations in autism. Nat. Genet. 2015, 47, 582–588.
- Almandil, N.B.; Alkuroud, D.N.; Abdulazeez, S.; Alsulaiman, A.; Elaissari, A.; Francis Borgio, J. Environmental and Genetic Factors in Autism Spectrum Disorders: Special Emphasis on Data from Arabian Studies. Int. J. Environ. Res. Public Health 2019, 16, 658.
- Chen, Y.; Liu, S.; Xu, H.; Zheng, H.; Bai, C.; Pan, W.; Zhou, H.; Liao, M.; Huang, C.; Dong, Q. Maternal exposure to low dose BDE209 and Pb mixture induced neurobehavioral anomalies in C57BL/6 male offspring. Toxicology 2019, 418, 70–80.
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G.; et al. Prevalence of autism spectrum disorder among children aged 8 Years-Autism and developmental disabilities monitoring network, 11 Sites, United States, 2016. MMWR Surveill. Summ. 2020, 69, 1–12.
- Leigh, J.P.; Du, J. Brief Report: Forecasting the Economic Burden of Autism in 2015 and 2025 in the United States. J. Autism Dev. Disord. 2015, 45, 4135–4139.
- Autism Statistics 2020: Is Autism an Epidemic? SingleCare. Available online: https://www.singlecare.com/blog/news/autism-statistics/ (accessed on 19 November 2020).
- Wing, L.; Gould, J. Severe impairments of social interaction and associated abnormalities in children: Epidemiology and classification. J. Autism Dev. Disord. 1979, 9, 11–29.
- Leary, M.R.; Hill, D.A. Moving on: Autism and movement disturbance. Ment. Retard. 1996, 34, 39–53.
- Shibutani, M.; Horii, T.; Shoji, H.; Morita, S.; Kimura, M.; Terawaki, N.; Miyakawa, T.; Hatada, I. Arid1b haploinsufficiency causes abnormal brain gene expression and autism-related behaviors in mice. Int. J. Mol. Sci. 2017, 18, 1872.
- Howsmon, D.P.; Kruger, U.; Melnyk, S.; James, S.J.; Hahn, J. Classification and adaptive behavior prediction of children with autism spectrum disorder based upon multivariate data analysis of markers of oxidative stress and DNA methylation. PLoS Comput. Biol. 2017, 13, e1005385.
- Chaidez, V.; Hansen, R.L.; Hertz-Picciotto, I. Gastrointestinal problems in children with autism, developmental delays or typical development. J. Autism Dev. Disord. 2014, 44, 1117–1127.
- Lord, C.; Brugha, T.; Charman, T.; Cusack, J.; Dumas, G.; Frazier, T.; Jones, E.; Jones, R.; Pickles, A.; State, M. Autism Spectrum Disorder. In Nature Reviews Disease Primers; Nature Publishing Group: London, UK, 2020; Volume 6, p. 5. ISBN 9780128093245.
- Kang, D.W.; Adams, J.B.; Gregory, A.C.; Borody, T.; Chittick, L.; Fasano, A.; Khoruts, A.; Geis, E.; Maldonado, J.; McDonough-Means, S.; et al. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: An open-label study. Microbiome 2017, 5, 10.
- Bakheet, S.A.; Alzahrani, M.Z.; Ansari, M.A.; Nadeem, A.; Zoheir, K.M.A.; Attia, S.M.; Al-Ayadhi, L.Y.; Ahmad, S.F. Resveratrol Ameliorates Dysregulation of Th1, Th2, Th17, and T Regulatory Cell-Related Transcription Factor Signaling in a BTBR T + tf/J Mouse Model of Autism. Mol. Neurobiol. 2017, 54, 5201–5212.
- Varghese, M.; Keshav, N.; Jacot-Descombes, S.; Warda, T.; Wicinski, B.; Dickstein, D.L.; Harony-Nicolas, H.; De Rubeis, S.; Drapeau, E.; Buxbaum, J.D.; et al. Autism spectrum disorder: Neuropathology and animal models. Acta Neuropathol. 2017, 134, 537–566.
- Li, Y.J.; Zhang, X.; Li, Y.M. Antineuroinflammatory therapy: Potential treatment for autism spectrum disorder by inhibiting glial activation and restoring synaptic function. CNS Spectr. 2019, 25, 493–501.
- Zamberletti, E.; Gabaglio, M.; Woolley-Roberts, M.; Bingham, S.; Rubino, T.; Parolaro, D. Cannabidivarin Treatment Ameliorates Autism-Like Behaviors and Restores Hippocampal Endocannabinoid System and Glia Alterations Induced by Prenatal Valproic Acid Exposure in Rats. Front. Cell. Neurosci. 2019, 13, 367.
- Rose, D.R.; Yang, H.; Serena, G.; Sturgeon, C.; Ma, B.; Careaga, M.; Hughes, H.K.; Angkustsiri, K.; Rose, M.; Hertz-Picciotto, I.; et al. Differential immune responses and microbiota profiles in children with autism spectrum disorders and co-morbid gastrointestinal symptoms. Brain. Behav. Immun. 2018, 70, 354–368.
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250.
- Katayama, Y.; Nishiyama, M.; Shoji, H.; Ohkawa, Y.; Kawamura, A.; Sato, T.; Suyama, M.; Takumi, T.; Miyakawa, T.; Nakayama, K.I. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nat. Publ. Gr. 2016, 537, 675–679.
- Bernier, R.; Golzio, C.; Xiong, B.; Stessman, H.A.; Coe, B.P.; Penn, O.; Witherspoon, K.; Gerdts, J.; Baker, C.; Vulto-Van Silfhout, A.T.; et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014, 158, 263–276.
- Talkowski, M.E.; Rosenfeld, J.A.; Blumenthal, I.; Pillalamarri, V.; Chiang, C.; Heilbut, A.; Ernst, C.; Hanscom, C.; Rossin, E.; Lindgren, A.M.; et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 2012, 149, 525–537.
- Blumenthal, I.; Ragavendran, A.; Erdin, S.; Klei, L.; Sugathan, A.; Guide, J.R.; Manavalan, P.; Zhou, J.Q.; Wheeler, V.C.; Levin, J.Z.; et al. Transcriptional consequences of 16p11.2 deletion and duplication in mouse cortex and multiplex autism families. Am. J. Hum. Genet. 2014, 94, 870–883.
- Wade, A.A.; Lim, K.; Catta-Preta, R.; Nord, A.S. Common CHD8 genomic targets contrast with model-specific transcriptional impacts of CHD8 haploinsufficiency. Front. Mol. Neurosci. 2019, 11, 481.
- Jiménez, J.A.; Ptacek, T.S.; Tuttle, A.H.; Schmid, R.S.; Moy, S.S.; Simon, J.M.; Zylka, M.J. Chd8 haploinsufficiency impairs early brain development and protein homeostasis later in life. Mol. Autism 2020, 11, 1–15.
- Wilkinson, B.; Grepo, N.; Thompson, B.L.; Kim, J.; Wang, K.; Evgrafov, O.V.; Lu, W.; Knowles, J.A.; Campbell, D.B. The autism-associated gene chromodomain helicase DNA-binding protein 8 (CHD8) regulates noncoding RNAs and autism-related genes. Transl. Psychiatry 2015, 5, e568.
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012, 338, 1619–1622.
- Li, J.; Wang, L.; Guo, H.; Shi, L.; Zhang, K.; Tang, M.; Hu, S.; Dong, S.; Liu, Y.; Wang, T.; et al. Targeted sequencing and functional analysis reveal brain-size-related genes and their networks in autism spectrum disorders. Mol. Psychiatry 2017, 22, 1282–1290.
- Douzgou, S.; Liang, H.W.; Metcalfe, K.; Somarathi, S.; Tischkowitz, M.; Mohamed, W.; Kini, U.; McKee, S.; Yates, L.; Bertoli, M.; et al. The clinical presentation caused by truncating CHD8 variants. Clin. Genet. 2019, 96, 72–84.
- Ostrowski, P.J.; Zachariou, A.; Loveday, C.; Beleza-Meireles, A.; Bertoli, M.; Dean, J.; Douglas, A.G.L.; Ellis, I.; Foster, A.; Graham, J.M.; et al. The CHD8 overgrowth syndrome: A detailed evaluation of an emerging overgrowth phenotype in 27 patients. Am. J. Med. Genet. Part C Semin. Med. Genet. 2019, 181, 557–564.
- An, Y.; Zhang, L.; Liu, W.; Jiang, Y.; Chen, X.; Lan, X.; Li, G.; Hang, Q.; Wang, J.; Gusella, J.F.; et al. De novo variants in the Helicase-C domain of CHD8 are associated with severe phenotypes including autism, language disability and overgrowth. Hum. Genet. 2020, 139, 499–512.
- Wang, T.; Guo, H.; Xiong, B.; Stessman, H.A.F.; Wu, H.; Coe, B.P.; Turner, T.N.; Liu, Y.; Zhao, W.; Hoekzema, K.; et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat. Commun. 2016, 7, 13316.
- Yasin, H.; Gibson, W.T.; Langlois, S.; Stowe, R.M.; Tsang, E.S.; Lee, L.; Poon, J.; Tran, G.; Tyson, C.; Wong, C.K.; et al. A distinct neurodevelopmental syndrome with intellectual disability, autism spectrum disorder, characteristic facies, and macrocephaly is caused by defects in CHD8. J. Hum. Genet. 2019, 64, 271–280.
- Stolerman, E.S.; Smith, B.; Chaubey, A.; Jones, J.R. CHD8 intragenic deletion associated with autism spectrum disorder. Eur. J. Med. Genet. 2016, 59, 189–194.
- Thompson, B.A.; Tremblay, V.; Lin, G.; Bochar, D.A. CHD8 Is an ATP-Dependent Chromatin Remodeling Factor That Regulates β-Catenin Target Genes. Mol. Cell. Biol. 2008, 28, 3894–3904.
- Nishiyama, M.; Oshikawa, K.; Tsukada, Y.-I.; Nakagawa, T.; Iemura, S.-I.; Natsume, T.; Fan, Y.; Kikuchi, A.; Skoultchi, A.I.; Nakayama, K.I. CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat. Cell Biol. 2009, 11, 172–182.
- Manning, B.J.; Yusufzai, T. The ATP-dependent chromatin remodeling enzymes CHD6, CHD7, and CHD8 exhibit distinct nucleosome binding and remodeling activities. J. Biol. Chem. 2017, 292, 11927–11936.
- Nishiyama, M.; Skoultchi, A.I.; Nakayama, K.I. Histone H1 Recruitment by CHD8 Is Essential for Suppression of the Wnt-Catenin Signaling Pathway. Mol. Cell. Biol. 2012, 3, 501–512.
- Subtil-Rodríguez, A.; Vázquez-Chávez, E.; Ceballos-Chávez, M.; Rodríguez-Paredes, M.; Martín-Subero, J.I.; Esteller, M.; Reyes, J.C. The chromatin remodeller CHD8 is required for E2F-dependent transcription activation of S-phase genes. Nucleic Acids Res. 2014, 42, 2185–2196.
- Ishihara, K.; Oshimura, M.; Nakao, M. CTCF-Dependent Chromatin Insulator Is Linked to Epigenetic Remodeling. Mol. Cell 2006, 23, 733–742.
- Batsukh, T.; Schulz, Y.; Wolf, S.; Rabe, T.I.; Oellerich, T.; Urlaub, H.; Schaefer, I.M.; Pauli, S. Identification and Characterization of FAM124B as a Novel Component of a CHD7 and CHD8 Containing Complex. PLoS ONE 2012, 7, e52640.
- Cotney, J.; Muhle, R.A.; Sanders, S.J.; Liu, L.; Willsey, A.J.; Niu, W.; Liu, W.; Klei, L.; Lei, J.; Yin, J.; et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat. Commun. 2015, 6, 6404.
- Zhao, C.; Dong, C.; Frah, M.; Deng, Y.; Marie, C.; Zhang, F.; Xu, L.; Ma, Z.; Dong, X.; Lin, Y.; et al. Dual Requirement of CHD8 for Chromatin Landscape Establishment and Histone Methyltransferase Recruitment to Promote CNS Myelination and Repair. Dev. Cell 2018, 45, 753–768.e8.
- Goodman, J.V.; Bonni, A. Regulation of neuronal connectivity in the mammalian brain by chromatin remodeling. Curr. Opin. Neurobiol. 2019, 59, 59–68.
- Durak, O.; Gao, F.; Kaeser-Woo, Y.J.; Rueda, R.; Martorell, A.J.; Nott, A.; Liu, C.Y.; Watson, L.A.; Tsai, L.H. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat. Neurosci. 2016, 19, 1477–1488.
- Sugathan, A.; Biagioli, M.; Golzio, C.; Erdin, S.; Blumenthal, I.; Manavalan, P.; Ragavendran, A.; Brand, H.; Lucente, D.; Miles, J.; et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4468–E4477.
- Sanders, S.J. First glimpses of the neurobiology of autism spectrum disorder. Curr. Opin. Genet. Dev. 2015, 33, 80–92.
- Kasah, S.; Oddy, C.; Basson, M.A. Autism-linked CHD gene expression patterns during development predict multi-organ disease phenotypes. J. Anat. 2018, 233, 755–769.
- Xu, Q.; Liu, Y.; Wang, X.; Tan, G.; Li, H.; Hulbert, S.W.; Li, C. Autism-associated CHD8 deficiency impairs axon development and migration of cortical neurons. Mol. Autism 2018, 9, 65.
- Jung, H.; Park, H.; Choi, Y.; Kang, H.; Lee, E.; Kweon, H.; Roh, J.D.; Ellegood, J.; Choi, W.; Kang, J.; et al. Sexually dimorphic behavior, neuronal activity, and gene expression in Chd8-mutant mice. Nat. Neurosci. 2018, 21, 1218–1228.
- Wang, P.; Mokhtari, R.; Pedrosa, E.; Kirschenbaum, M.; Bayrak, C.; Zheng, D.; Lachman, H.M. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol. Autism 2017, 8, 11.
- Sood, S.; Weber, C.M.; Hodges, H.C.; Krokhotin, A.; Shalizi, A.; Crabtree, G.R. CHD8 dosage regulates transcription in pluripotency and early murine neural differentiation. Proc. Natl. Acad. Sci. USA 2020, 117, 22331–22340.
- Siu, M.T.; Butcher, D.T.; Turinsky, A.L.; Cytrynbaum, C.; Stavropoulos, D.J.; Walker, S.; Caluseriu, O.; Carter, M.; Lou, Y.; Nicolson, R.; et al. Functional DNA methylation signatures for autism spectrum disorder genomic risk loci: 16p11.2 deletions and CHD8 variants. Clin. Epigenetics 2019, 11, 103.
- Wong, W.R.; Brugman, K.I.; Maher, S.; Oh, J.Y.; Howe, K.; Kato, M.; Sternberg, P.W. Autism-Associated missense genetic variants impact locomotion and neurodevelopment in Caenorhabditis elegans. Hum. Mol. Genet. 2019, 28, 2271–2281.
- Genç, Ö.; An, J.Y.; Fetter, R.D.; Kulik, Y.; Zunino, G.; Sanders, S.J.; Davis, G.W. Homeostatic plasticity fails at the intersection of autism-gene mutations and a novel class of common genetic modifiers. Elife 2020, 9, e55775.
- Coll-Tané, M.; Gong, N.N.; Belfer, S.J.; Renssen, L.v.; Kurtz-Nelson, E.C.; Szuperak, M.; Eidhof, I.; Reijmersdal, B.v.; Terwindt, I.; Verheij, M.M.; et al. The CHD8/CHD7/Kismet family links blood-brain barrier glia and serotonin to ASDassociated sleep defects. Sci. Adv. 2021, 7, eabe2626.
- Kita, Y.; Katayama, Y.; Shiraishi, T.; Oka, T.; Sato, T.; Suyama, M.; Ohkawa, Y.; Miyata, K.; Oike, Y.; Shirane, M.; et al. The Autism-Related Protein CHD8 Cooperates with C/EBPβ to Regulate Adipogenesis. Cell Rep. 2018, 23, 1988–2000.
- Marie, C.; Clavairoly, A.; Frah, M.; Hmidan, H.; Yan, J.; Zhao, C.; Van Steenwinckel, J.; Daveau, R.; Zalc, B.; Hassan, B.; et al. Oligodendrocyte precursor survival and differentiation requires chromatin remodeling by Chd7 and Chd8. Proc. Natl. Acad. Sci. USA 2018, 115, E8246–E8255.
- Cherepanov, S.M.; Gerasimenko, M.; Yuhi, T.; Furuhara, K.; Tsuji, C.; Yokoyama, S.; Nakayama, K.I.; Nishiyama, M.; Higashida, H. Oxytocin ameliorates impaired social behavior in a Chd8 haploinsufficiency mouse model of autism. BMC Neurosci. 2021, 22, 32.
- Platt, R.J.; Zhou, Y.; Slaymaker, I.M.; Shetty, A.S.; Weisbach, N.R.; Kim, J.A.; Sharma, J.; Desai, M.; Sood, S.; Kempton, H.R.; et al. Chd8 Mutation Leads to Autistic-like Behaviors and Impaired Striatal Circuits. Cell Rep. 2017, 19, 335–350.
- Gompers, A.L.; Su-Feher, L.; Ellegood, J.; Copping, N.A.; Riyadh, M.A.; Stradleigh, T.W.; Pride, M.C.; Schaffler, M.D.; Wade, A.A.; Catta-Preta, R.; et al. Germline Chd8 haploinsufficiency alters brain development in mouse. Nat. Neurosci. 2017, 20, 1062–1073.
- Suetterlin, P.; Hurley, S.; Mohan, C.; Riegman, K.L.H.; Pagani, M.; Caruso, A.; Ellegood, J.; Galbusera, A.; Crespo-Enriquez, I.; Michetti, C.; et al. Altered neocortical gene expression, brain overgrowth and functional over-connectivity in chd8 haploinsufficient mice. Cereb. Cortex 2018, 28, 2192–2206.
- Kawamura, A.; Katayama, Y.; Nishiyama, M.; Shoji, H.; Tokuoka, K.; Ueta, Y.; Miyata, M.; Isa, T.; Miyakawa, T.; Hayashi-Takagi, A.; et al. Oligodendrocyte dysfunction due to Chd8 mutation gives rise to behavioral deficits in mice. Hum. Mol. Genet. 2020, 29, 1274–1291.
- Hulbert, S.W.; Wang, X.; Gbadegesin, S.O.; Xu, Q.; Xu, X.; Jiang, Y.H. A Novel Chd8 Mutant Mouse Displays Altered Ultrasonic Vocalizations and Enhanced Motor Coordination. Autism Res. 2020, 13, 1685–1697.
- Kawamura, A.; Katayama, Y.; Kakegawa, W.; Ino, D.; Nishiyama, M.; Yuzaki, M.; Nakayama, K.I. The autism-associated protein CHD8 is required for cerebellar development and motor function. Cell Rep. 2021, 35.
- Phelan, K.; Rogers, R.C.; Boccuto, L. Phelan-McDermid Syndrome. In GeneReviews®; University of Washington: Seattle, WA, USA, 2018.
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215.
- Gregor, A.; Oti, M.; Kouwenhoven, E.N.; Hoyer, J.; Sticht, H.; Ekici, A.B.; Kjaergaard, S.; Rauch, A.; Stunnenberg, H.G.; Uebe, S.; et al. De novo mutations in the genome organizer CTCF cause intellectual disability. Am. J. Hum. Genet. 2013, 93, 124–131.
- Shingleton, J.R.; Hemann, M.T. The chromatin regulator CHD8 is a context-dependent mediator of cell survival in murine hematopoietic malignancies. PLoS ONE 2015, 10, e0143275.
- Orly Weissberg; Evan Elliott; The Mechanisms of CHD8 in Neurodevelopment and Autism Spectrum Disorders. Genes 2021, 12, 1133, 10.3390/genes12081133.


