 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Svetlana Sharifulina | + 1378 word(s) | 1378 | 2021-08-06 11:14:23 | | | |
| 2 | Amina Yu | Meta information modification | 1378 | 2021-08-19 09:47:31 | | |
Video Upload Options
The transcription factors and signaling proteins that play an important role in brain cell responses to ischemia undergo acetylation/deacetylation. In different cell types, the acetylation/deacetylation of different regions of non-histone proteins containing lysines occurs and HAT/HDAC activity depends on the acetylation site. Moreover, the activity of the HDACs themselves can be regulated by their acetylation/deacetylation.
1. Introduction
Acetylation of histones and non-histone proteins modulates gene expression and signaling in cells. Changes in the secondary structure of proteins by acetylation leads to a change in their enzymatic activity, subcellular localization and protein-protein interactions [1].
The study of non-histone protein acetylation/deacetylation began after the success of the clinical use of histone deacetylase inhibitors (HDACs) in the treatment of various forms of cancer and was driven by the search for the causes of cytotoxicity of nonselective HDAC inhibitors (HDACi) [2]. Non-histone substrates of HDAC and acetyltransferases (HAT) have been identified, which are tumor suppressors, signaling mediators, steroid receptors and transcription factors [3][4]. The number of identified proteins in which activity is regulated by acetylation/deacetylation to date is certainly lower than the actual amount represented by acetylome in vivo.
There are very few studies on the non-histone protein acetylation/deacetylation in brain cells, and there is apparently no data on these processes in stroke. Our review aims to analyze the possible role of acetylation/deacetylation of transcription factors and signaling proteins involved in the regulation of apoptosis in ischemia.
2. Protein Acetylation and Deacetylation Enzymes
Acetylation and deacetylation of histones and non-histone proteins is carried out by histone deacetylase (HDAC) and histone acetyltransferase (HAT). Histone acetyltransferases (HATs) transfer acetyl groups from acetyl coenzyme A (Acetyl Co-A) to the ε-amino group of lysine residues, while histone deacetylases (HDACs), on the contrary, catalyze the removal of acetyl groups. Since histones were the first identified targets of deacetylases and acetyltransferases, these enzymes were named histone deacetylases and histone acetyltransferases. However, in addition to regulating transcription, HAT/HDAC regulate the most important functions of cells by acetylating/deacetylating a huge number of non-histone proteins, which have always been their evolutionarily primary targets [3]. These proteins regulate cell survival and death, replication, DNA repair, the cell cycle, and cell responses to stress and aging.
3. c-Myc
One of the main regulators of many target genes is the transcription factor c-Myc. It activates (or sometimes suppresses) 10–15% of all genes involved in the regulation of energy metabolism, protein synthesis, oncogenesis, the cell cycle, and apoptosis. It functions at both the transcriptional and epigenetic level. It can potentiate apoptosis. c-Myc is an oncogene in many human tumors [5]. c-Myc regulation can be involved in several signaling pathways, such as JAK/STAT, Wnt/β-catenin, Notch, and the Ras/PI3K/AKT/GSK-3 signaling pathways, that increase c-Myc levels [6].
Its overexpression was also noted after transient global or focal cerebral ischemia in rodents, where c-Myc promoted neuronal death [7]. An increase in the level of c-Myc was observed in the penumbra on the first day after PTS [8].
The stability of c-Myc in different types of cancer cells is associated with its acetylation at lysine 323 by PCAF acetyltransferase ( Figure 1 ) [9]. In lymphoma cells, SIRT1 interacted with the C-terminus of c-Myc and deacetylated it both in vitro and in vivo [10]. However, inhibitors of HDAC, but not sirtuins, increased the acetylation of c-Myc at lysine 323 and inhibited tumorigenesis [11], which promoted the association of c-Myc with Max, a partner required for c-Myc activation. HDAC inhibitors downregulated c-Myc by blocking GSK-3 phosphorylation and exhibited synergistic cytotoxic and c-Myc-suppressive effects ( Figure 2 ) [12]. HDAC3 also deacetylated c-Myc at lysine 323 in cholangiocarcinoma cells, which protected the protein from ubiquitin-dependent proteolysis [13]. Thus, it can be assumed that at least one of the HDACs in brain cells is c-Myc deacetylase. c-Myc stimulates the expression of p53 and E2F1 [14].
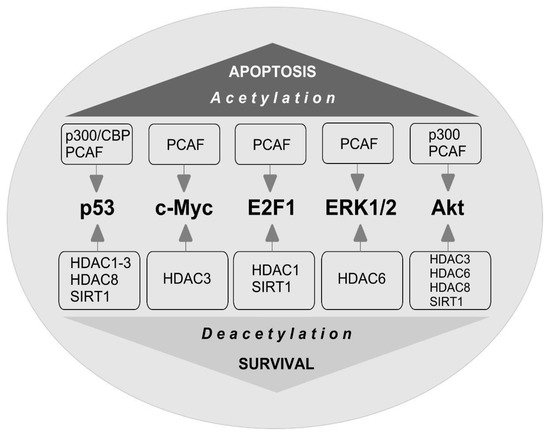
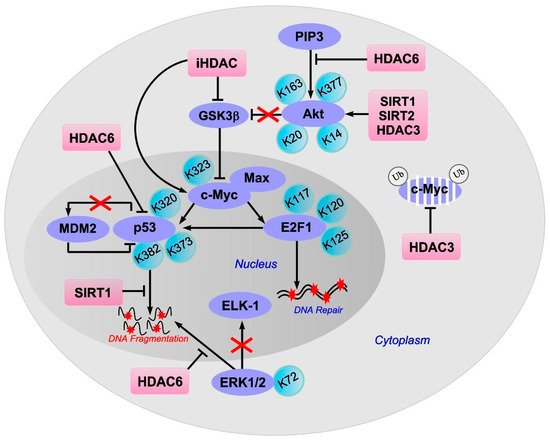
4. E2F1
E2F1 transcription factor is one of the key players in determining the fate of the cell. It controls the expression of various genes that regulate DNA synthesis and repair, the cell cycle, and apoptosis [15][16]. E2F1 stimulates apoptosis when the cell cycle is disrupted or suppressed, which is typical of neurons [17]. Its synthesis is controlled by the p38 MAP kinase and the c-Myc transcription factor [18]. E2F1 induces the expression of various proapoptotic proteins, such as caspases 3, 7, 8, and 9, Smac/DIABLO, Apaf-1, Bcl-2, p53, and p73 [17][19]. Overexpression of E2F1, p53, c-Myc, p38, Smac/DIABLO, Bcl-x, caspases 3, 6, and 7 was observed in the penumbra on the first day after PTS [8]. This is consistent with data on the increased expression of E2F1 [20] and p53 [21] in the penumbra after MCAO and in the axotomized spinal ganglia of rats [22]. Inhibition of the E2F1/p53 pathway prevents neuronal apoptosis [19].
iHDACs have been shown to affect E2F1 activity [23]. In cancer cells, in response to genotoxic stress caused by doxorubicin, E2F1 is acetylated by PCAF at three lysines (K117, 120, and 125). This stabilizes the protein and increases its specific binding to DNA [24][25]. Acetylation of these lysines induces the accumulation of ubiquitinated but stable E2F1 [26]. Acetylation of E2F1 promotes the recruitment of chromatin-modifying enzymes and DNA double-strand break repair factors [27].
HDAC1 acts as a deacetylase in different types of cancer cells [24][28][29]. In retinal epithelial cells, E2F1 is deacetylated by Sirt1, which contributes to the resistance of cells to oxidative stress caused by H2O2 [30]. Thus, acetylation/deacetylation of E2F1 can contribute to the resistance of different types of cells to damage. However, in the literature available to us, we failed to find information on the acetylation/deacetylation of E2F1 in brain cells, either in normal conditions or in pathology.
References
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and Functional Diversity of Lysine Acetylation Revealed by a Proteomics Survey. Mol. Cell 2006, 23, 607–618.
- Lakshmaiah, K.C.; Jacob, L.A.; Aparna, S.; Lokanatha, D.; Saldanha, S.C. Epigenetic therapy of cancer with histone deacetylase inhibitors. J. Cancer Res. Ther. 2014, 10, 469–478.
- Spange, S.; Wagner, T.; Heinzel, T.; Krämer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198.
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. p53 at the Crossroads between Different Types of HDAC Inhibitor-Mediated Cancer Cell Death. Int. J. Mol. Sci. 2019, 20, 2415.
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990.
- Chanvorachote, P.; Sriratanasak, N.; Nonpanya, N. C-myc Contributes to Malignancy of Lung Cancer: A Potential Anticancer Drug Target. Anticancer. Res. 2020, 40, 609–618.
- McGahan, L.; Hakim, A.M.; Robertson, G.S. Hippocampal Myc and p53 expression following transient global ischemia. Mol. Brain Res. 1998, 56, 133–145.
- Demyanenko, S.; Uzdensky, A. Profiling of Signaling Proteins in Penumbra After Focal Photothrombotic Infarct in the Rat Brain Cortex. Mol. Neurobiol. 2017, 54, 6839–6856.
- Patel, J.H.; Du, Y.; Ard, P.G.; Phillips, C.; Carella, B.; Chen, C.-J.; Rakowski, C.; Chatterjee, C.; Lieberman, P.M.; Lane, W.S.; et al. The c-MYC Oncoprotein Is a Substrate of the Acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 2004, 24, 10826–10834.
- Mao, B.; Zhao, G.; Lv, X.; Chen, H.-Z.; Xue, Z.; Yang, B.; Liu, D.-P.; Liang, C.-C. Sirt1 deacetylates c-Myc and promotes c-Myc/Max association. Int. J. Biochem. Cell Biol. 2011, 43, 1573–1581.
- Nebbioso, A.; Carafa, V.; Conte, M.; Tambaro, F.P.; Abbondanza, C.; Martens, J.; Nees, M.; Benedetti, R.; Pallavicini, I.; Minucci, S.; et al. c-Myc Modulation and Acetylation Is a Key HDAC Inhibitor Target in Cancer. Clin. Cancer Res. 2017, 23, 2542–2555.
- Ecker, J.; Thatikonda, V.; Sigismondo, G.; Selt, F.; Valinciute, G.; Oehme, I.; Müller, C.; Buhl, J.L.; Ridinger, J.; Usta, D.; et al. Reduced chromatin binding of MYC is a key effect of HDAC inhibition in MYC amplified medulloblastoma. Neuro-Oncology 2021, 23, 226–239.
- Zhang, M.; Pan, Y.; Tang, D.; Dorfman, R.G.; Xu, L.; Zhou, Q.; Zhou, L.; Wang, Y.; Li, Y.; Yin, Y.; et al. Low levels of pyruvate induced by a positive feedback loop protects cholangiocarcinoma cells from apoptosis. Cell Commun. Signal. 2019, 17, 1–14.
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776.
- Meng, P.; Ghosh, R. Transcription addiction: Can we garner the Yin and Yang functions of E2F1 for cancer therapy? Cell Death Dis. 2014, 5, e1360.
- Mathey-Prevot, B.; Parker, B.-T.; Im, C.; Hong, C.; Dong, P.; Yao, G.; You, L. Quantifying E2F1 protein dynamics in single cells. Quant. Biol. 2020, 8, 20–30.
- Folch, J.; Junyent, F.; Verdaguer, E.; Auladell, C.; Pizarro, J.G.; Beas-Zárate, C.; Pallàs, M.; Camins, A. Role of Cell Cycle Re-Entry in Neurons: A Common Apoptotic Mechanism of Neuronal Cell Death. Neurotox. Res. 2011, 22, 195–207.
- Bretones, G.; Delgado, M.D.; León, J. Myc and cell cycle control. Biochim. Biophys. Acta-Gene Regul. Mech. 2015, 1849, 506–516.
- Camins, A.; Verdaguer, E.; Folch, J.; Beas-Zarate, C.; Canudas, A.M.; Pallas, M. Inhibition of Ataxia Telangiectasia-p53-E2F-1 Pathway in Neurons as a Target for the Prevention of Neuronal Apoptosis. Curr. Drug Metab. 2007, 8, 709–715.
- MacManus, J.P.; Jian, M.; Preston, E.; Rasquinha, I.; Webster, J.; Zurakowski, B. Absence of the Transcription Factor E2F1 Attenuates Brain Injury and Improves Behavior after Focal Ischemia in Mice. J. Cereb. Blood Flow Metab. 2003, 23, 1020–1028.
- Li, Y.; Chopp, M.; Zhang, Z.G.; Zaloga, C.; Niewenhuis, L.; Gautam, S. p53-immunoreactive protein and p53 mRNA expression after transient middle cerebral artery occlusion in rats. Stroke 1994, 25, 849–855.
- Dzreyan, V.; Rodkin, S.; Nikul, V.; Pitinova, M.; Uzdensky, A. The Expression of E2F1, p53, and Caspase 3 in the Rat Dorsal Root Ganglia After Sciatic Nerve Transection. J. Mol. Neurosci. 2021, 71, 826–835.
- Abramova, M.V.; Pospelova, T.V.; Nikulenkov, F.P.; Hollander, C.M.; Fornace, A.J.; Pospelov, V.A. G1/S Arrest Induced by Histone Deacetylase Inhibitor Sodium Butyrate in E1A + Ras-transformed Cells Is Mediated through Down-regulation of E2F Activity and Stabilization of β-Catenin. J. Biol. Chem. 2006, 281, 21040–21051.
- Martínez-Balbás, M.; Bauer, U.-M.; Nielsen, S.J.; Brehm, A.; Kouzarides, T. Regulation of E2F1 activity by acetylation. EMBO J. 2000, 19, 662–671.
- Ianari, A.; Gallo, R.; Palma, M.; Alesse, E.; Gulino, A. Specific Role for p300/CREB-binding Protein-associated Factor Activity in E2F1 Stabilization in Response to DNA Damage. J. Biol. Chem. 2004, 279, 30830–30835.
- Galbiati, L.; Mendoza-Maldonado, R.; Gutierrez, M.I.; Giacca, M. Regulation of E2F-1 after DNA Damage by p300-Mediated Acetylation and Ubiquitination. Cell Cycle 2005, 4, 930–939.
- Xia, C.; Tao, Y.; Li, M.; Che, T.; Qu, J. Protein acetylation and deacetylation: An important regulatory modification in gene transcription (Review). Exp. Ther. Med. 2020, 20, 2923–2940.
- Zhang, W.; Ji, W.; Liu, X.; Ouyang, G.; Xiao, W. ELL Inhibits E2F1 Transcriptional Activity by Enhancing E2F1 Deacetylation via Recruitment of Histone Deacetylase 1. Mol. Cell. Biol. 2014, 34, 765–775.
- Wu, M.; Seto, E.; Zhang, J. E2F1 enhances glycolysis through suppressing Sirt6 transcription in cancer cells. Oncotarget 2015, 6, 11252–11263.
- Gong, C.; Qiao, L.; Feng, R.; Xu, Q.; Zhang, Y.; Fang, Z.; Shen, J.; Li, S. IL-6-induced acetylation of E2F1 aggravates oxidative damage of retinal pigment epithelial cell line. Exp. Eye Res. 2020, 200, 108219.

