 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sandrine Herbelet | + 1916 word(s) | 1916 | 2020-07-12 11:11:20 | | | |
| 2 | Nicole Yin | + 160 word(s) | 2076 | 2020-07-16 11:11:13 | | | | |
| 3 | Nicole Yin | -23 word(s) | 2053 | 2020-11-10 03:09:56 | | |
Video Upload Options
In Duchenne muscular dystrophy (DMD), the activation of proinflammatory and metabolic cellular pathways in skeletal muscle cells is an inherent characteristic. Synthetic glucocorticoid intake counteracts the majority of these mechanisms. However, glucocorticoids induce burdensome secondary effects, including hypertension, arrhythmias, hyperglycemia, osteoporosis, weight gain, growth delay, skin thinning, cushingoid appearance, and tissue-specific glucocorticoid resistance. Hence, lowering the glucocorticoid dosage could be beneficial for DMD patients. A more profound insight into the major cellular pathways that are stabilized after synthetic glucocorticoid administration in DMD is needed when searching for the molecules able to achieve similar pathway stabilization. This review provides a concise overview of the major anti-inflammatory pathways, as well as the metabolic effects of glucocorticoids in the skeletal muscle affected in DMD. The known drugs able to stabilize these pathways, and which could potentially be combined with glucocorticoid therapy as steroid-sparing agents, are described. This could create new opportunities for testing in DMD animal models and/or clinical trials, possibly leading to smaller glucocorticoids dosage regimens for DMD patients.
1. Definition
Duchenne muscular dystrophy (DMD) is an X-linked recessive skeletal muscle disorder affecting around one in 3500 boys. It is characterized by muscle weakness and wasting with a clinical onset at around 4 years of age.
2. Introduction
The absence of dystrophin, the protein providing a link between the extracellular matrix and the myocyte cytoskeleton, destabilizes the dystrophin-associated protein complex (DAPC), a large transmembrane protein complex that acts as a shock absorber during muscle contraction, a receiver and transducer of cellular signals and as a scaffold for signaling proteins[1][2]. Abnormal shock absorption is thought to result in increased sarcolemmal damage in the myocyte during exercise. Aberrant signal transduction in DMD is believed to result from the disturbed link between neuronal nitric oxide synthase (nNOS), dystrophin, and the DAPC. Stretch-activated channels are located downstream of dystrophin that lead to the activation of the extracellular signal regulated kinase-mitogen activated protein kinase (ERK-MAPK) cascade, along with increased Ca2+ influx, in DMD. Together, these pathological mechanisms induce disorganized muscle with myofiber hypertrophy and necrosis, inflammation, fibrosis, and fat deposition[3][4][5].
Administration of glucocorticoids (GCs) can counteract these phenomena in patients with DMD. Different GC dosing regimens can be used. The most commonly used consists of an intake of 0.75 mg/kg/day of prednisone. This is based on the strength and functional outcomes in randomized controlled trials (RCTs), as summarized in the systematic review of Matthews et al.[6]. Deflazacort is another commonly used GC. These GCs prolong ambulation and mildly improve muscle strength. The major effect of GCs is believed to reside in their inhibition of the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-ƘB), being the master inducer of inflammation. Unfortunately, GCs can induce several side-effects, such as suppression of the immune system, potentially leading to life-threatening infections, arrhythmias, hypertension, hyperglycemia and glucosuria, cataracts, increased food intake and weight gain, delayed growth, osteoporosis and fractures, abnormal behavior, insomnia, cushingoid appearance, easy bruising, gastro-intestinal symptoms, and hirsutism[6][7].
GCs regulate reproduction, development, cell differentiation, inflammation, and responses of the immune system[8]. GCs bind in the cytoplasm to the glucocorticoid receptor (GR), also known as Nuclear Receptor Subfamily 3 Group C Member 1 (NR3C1). Two isoforms termed GRα and GRβ have been described. These are the two main transcriptional isoforms[8][9][10]. While unstimulated, the GR resides mainly in the cytoplasm and is chaperoned by two heat shock protein 90 (Hsp90) molecules, Hsp90-binding protein 23 (p23), Hsp70, and immunophilins, all converging to protect the GR from degradation and prepare it for GC binding at the receptor site. Binding to the GR releases the Hsps[8][9][10]. The GR can translocate from the cytoplasm to the nucleus and back, sensing the intracellular environment along its journey and interacting with several proteins, thereby creating the GR interactome. After the binding of GCs to the GR, the actions of the transcription factor can be divided in genomic and non-genomic functions[8][9][10][11]. One of the major targets of the GR is NF-ƘB, the major activator of the genes coding for immunoreceptors, proinflammatory cytokines, and chemokines along with the other important cellular players inhibited by GCs. Genomic effects appear after GC–GR translocation to the nucleus and subsequent DNA binding, and can result in expression changes in up to 10–20% of genes[10]. Non-genomic actions occur within minutes and consist of the activation of Toll-like receptors (TLRs), p38 mitogen-activated protein kinases (MAPKs), c-Jun N-terminal kinases (JNKs), the NF-ƘB and activator protein 1 (AP-1), the Janus kinase/signal transducer and activator of transcription proteins (JAK/STAT), and transforming growth factor-β (TGF-β).
In this review, the current evidence on the cellular pathways that are activated in and interact with GCs in the DMD myocyte, is summarized and discussed. The impact of glucocorticoid administration is visualized, thereby showing the normalization or exacerbation of the triggered pathways. Importantly, the purpose of this review is to discuss putative approaches to downscale the GC dosage in DMD patients via supplementation of the pathway stabilizing agents. All ongoing clinical trials for DMD in the USA can be monitored here: https://www.clinicaltrials.gov and in Europe on this site: https://www.clinicaltrialsregister.eu/.
3. Latest Research
When considering a polypharmaceutical approach for DMD patients, it is of the utmost importance to know which molecules should never be combined with GCs. Cytochrome P450 3A4 (CYP3A4) inhibitors, such as cobicistat (Gilead Sciences, Foster City, CA, USA), should not be administered alongside GCs[12]. The combination of GCs with glucose lowering drugs, such as metformin, requires great caution. It should be kept in mind that myostatin seems to be associated with the GCs induced catabolic effect on muscle proteins[13]and HDACis can cause severe side-effects, especially when combined with VPA[14]. In the past, a high dosage regimen of halofuginone, an anti-parasitic molecule used in veterinary medicine, combined with GCs led to the death of a DMD patient and abrogation of the clinical trial in 2016[15]. One possible explanation could be found in the synergy between halofuginone and GCs[16][17][18][19]. When considering polypharmacy in DMD, exploring molecular interactions is of great importance in avoiding life-threatening drug combinations.
From the therapeutic armamentarium tested in mdx mice and in DMD patients, the vast majority are molecules which were primarily designed for other diseases such as RA, Alzheimer’s disease, and regulation of the blood pressure or glucose homeostasis, to name a few. When blocking an entire pathway, such as the TGF-β, TLR, JAK/STAT or the glucose metabolism pathway, many other functions in the cell will be affected with repercussions in the cellular activity, which can be noticed as side-effects in patients (Tables 1-8). Inhibition of the TGF-β pathway seems to often produce side-effects (Table 5)[20][18][19][21][22]. Although some molecules have not yet been tested in clinical trials with DMD patients, such as losartan, suramin, and decorin, the potential side-effects make these molecules less attractive. The TGF-β cellular pathway is involved in many cellular functions ranging from cell growth to apoptosis and should be considered as a cellular ‘highway’ which upon blocking is likely to yield side-effects. Therefore, anti-TGF-β signaling drugs are challenging in finding a therapeutic window and dosing regimen still efficacious and with only minor side effects[23]. Follistatin is an autocrine glycoprotein expressed on nearly all cells of the body and side-effects were noticed during the Phase I/II trial (NCT02354781). Inhibition of myostatin, a myokine, is muscle specific. Usage of antibodies against myostatin will reveal in the near future their therapeutic value. The first outcome of the Phase II (NCT02606136) clinical trial currently ongoing in the USA, should be known after completion of the trial in April 2021.
Although the TLR-pathway is more oriented on the inflammatory response in skeletal muscle[24], many downstream proteins have broad cellular function, especially MAPK, which plays an important role in cellular proliferation[25]. From the TLR inhibitors listed as putative stabilizing agents, NI-0101 was used in RA in a Phase II clinical trial and did not show any improvement in the disease parameters[26]. Therefore, it may be less interesting to focus on this antibody. When considering miRNAs as a therapeutic option in DMD, cautiousness is required as miRNAs can harbor several modes of action. miR-146a deficiency in mdx mice did not aggravate the phenotype, but a slight increase in muscle damage and inflammation was observed[27]. Therefore, inhibiting miR-146a should be considered very carefully. Caution is also required when considering miR-21 inhibition. Indeed, in mdx mice, miR-21 is increased in fibroblasts and downregulated in myoblasts[28] [19], presumably making it a difficult target. miR-206 is known for activating a large array of proteins such as HDAC4, PTB, utrophin, Fstl1, Cx43, and TIMP3 and for inhibiting IGF-1, Pax3, and Pax7[29]. Despite promising results in mdx mice with (ALA)/L-carnitine (L-car) and its FDA approval, the absence of palmitoyl carnitine hydrolase in DMD muscle and its essential role in carnitine metabolism, makes this molecule uninteresting to consider as a stabilizing agent as it did not show any difference in function of extremities in DMD patients[30][31][32]. Considering the JNK inhibitor JIP1 is less interesting as it is generally administered through adenoviral infection, which makes it less suitable. The NF-κB inhibitors PDTC and IRFI-042 and the AP-1 inhibitor batimastat (Table 3) can all be orally ingested. However, batimastat displays a low oral bioavailability[33]. Neither PDTC nor IRFI-042 are FDA approved and could therefore not be considered for clinical trials in conjunction with GCs[34][35][36][37][38][39][40][41][42]. The JAK/STAT pathway stabilizing agent VPA, on the other hand, is FDA approved (ANDA 073229). It activates the PI3K/Akt/mTOR pathway and reduces apoptosis. It may be worth considering for DMD clinical trials yet keeping in mind it should never be combined with HDACis. This combination can trigger status epilepticus[43][14]. Potential danger for DMD patients could also arise when combining glucose lowering molecules with GCs, which should be avoided at any time (Table 6). Less dangerous, but still detrimental, are the side-effects seen with HDACis (Table 7). With arrhythmias, nausea, vomiting, anorexia, fatigue, anemia, and liver toxicities having been described, HDACis should be considered very cautiously in DMD[14]. Therefore, it is understandable that the scientific community has high hopes for vamorolone. Nevertheless, the cross-talk between the inflammatory and metabolic pathways through p38 MAPK, mTOR, and Akt, as depicted in Figure 1, could lead to the combination of several stabilizing molecules in conjunction with GCs and a subsequent decrease in GC-dosing. The theoretical combination of GCs with VPA should first be tested in mdx mice. If beneficial effects are observed, a polypharmaceutical approach could be applied in a clinical trial, potentially leading to smaller GC regimens for DMD patients, which would be beneficial for their health.
Many research efforts are put in gene and cell therapies for DMD. To name some gene therapies: exon skipping, stop codon readthrough, AAV-mediated therapies, RNA interference, clustered regularly interspaced short palindromic repeats (CRISPR/Cas9), all with the intention to restore dystrophin production in the DMD myocyte[44][45][46][47][48]. Cell therapies consist of autologous or allogenic muscle stem cell implantation in DMD patients[49]. A mixed approach of gene therapy with cell therapy along with GCs intake and supplementation of a steroid sparing agent such as VPA, could be worth considering. The cellular perturbations due to the lack of dystrophin and instable DAPC need to be addressed along with restoring dystrophin expression by gene therapy. Polypharmacy in DMD should always be applied after careful study of potentially life-threatening side-effects when combining several molecules, to safeguard the life of our DMD patients.
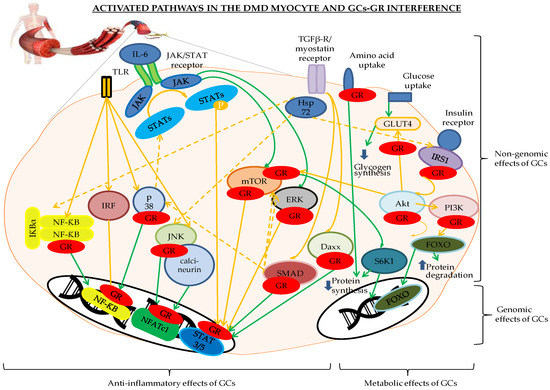
Figure 1. Genomic and non-genomic effects of glucocorticoid receptor (GR) binding to the different players of the inflammatory and metabolic pathways involved in the Duchenne muscular dystrophy (DMD) myocyte. The pathways indicated in orange are activated, the green indicates a return to the physiological status. The GR is depicted in red. Abbreviations: Daxx = death-associated protein, FOXO = forkhead box O, GLUT4 = glucose transporter 4, GR = glucocorticoid receptor, IκB = inhibitor of NF-κB, IRFs = interferon regulatory factors, Hsp72 = heat shock protein 72, IRS1 = insulin receptor substrate 1, JAK/STAT = Janus kinase/signal transducer and activator of transcription proteins, JNK = c-Jun N-terminal kinase, NFATc1 = nuclear translocation of nuclear factor of activated T cells c1, NF-κB = nuclear factor kappa-light-chain-enhancer of activated B cells, P = phosphorylation, p38 = p38 mitogen-activated protein kinases, PI3K/Akt/mTOR = Phosphoinositide 3-kinases/protein kinase B/mammalian target of rapamycin pathway, RAF/RAS/ERK = Rapidly Accelerated Fibrosarcoma/Ras-extracellular signal-regulated kinase, S6K1 = ribosomal protein S6 kinase beta-1, SMAD3 and -6 = Mothers Against Decapentaplegic homolog 3 and 6, TGF-β = transforming growth factor-β, TLR= Toll-like receptors.
References
- Fedik Rahimov; L M Kunkel; Cellular and molecular mechanisms underlying muscular dystrophy. The Journal of Cell Biology 2013, 201, 499-510, 10.1083/jcb.201212142.
- Bruno Constantin; Dystrophin complex functions as a scaffold for signalling proteins. Biochimica et Biophysica Acta (BBA) - Biomembranes 2014, 1838, 635-642, 10.1016/j.bbamem.2013.08.023.
- Simon Guiraud; Annemieke Aartsma-Rus; Natassia M. Vieira; Kay E. Davies; Gert-Jan B. Van Ommen; Louis M. Kunkel; The Pathogenesis and Therapy of Muscular Dystrophies. Annual Review of Genomics and Human Genetics 2015, 16, 281-308, 10.1146/annurev-genom-090314-025003.
- Nicolas Deconinck; Bernard Dan; Pathophysiology of Duchenne Muscular Dystrophy: Current Hypotheses. Pediatric Neurology 2007, 36, 1-7, 10.1016/j.pediatrneurol.2006.09.016.
- Kristin Wilson; Crystal Faelan; Janet Patterson-Kane; Daniel G. Rudmann; Steven A. Moore; Diane Frank; Jay Charleston; Jon Tinsley; G. David Young; Anthony J. Milici; et al. Duchenne and Becker Muscular Dystrophies: A Review of Animal Models, Clinical End Points, and Biomarker Quantification.. Toxicologic Pathology 2017, 45, 961-976, 10.1177/0192623317734823.
- Emma Matthews; Ruth Brassington; Thierry Kuntzer; Fatima Jichi; Adnan Y Manzur; Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database of Systematic Reviews 2016, 5, CD003725, 10.1002/14651858.cd003725.pub4.
- Valeria Ricotti; Deborah A Ridout; Elaine Scott; Ros Quinlivan; Stephanie A Robb; Adnan Y. Manzur; Francesco Muntoni; Adnan Y. Manzur; Francesco Muntoni; S. Robb; et al.Ros QuinlivanValeria RicottiM. MainK. BushbyV. StraubA. SarkozyM. GuglieriE. StrehleM. EagleA. MayhewHelen RoperH. McMURCHIEA. ChildsK. PysdenL. PallantS. SpintyG. PeacheyA. ShillingtonE. WraigeHeinz JungbluthJ. SheehanR. SpahrI. HughesE. BatemanC. CammissT. WillisL. GrovesN. EmeryP. BaxterM. SeniorL. HartleyB. ParsonsA. MajumdarL. JenkinsK. NaismithA. KeddieI. HorrocksM. Di MarcoG. ChowA. MiahC. De GoedeN. ThomasM. GearyJ. PalmerC. WhiteK. GreenfieldElaine Scotton behalf of the NorthStar Clinical NetworkGreat Ormond Street Hospital For Children Nhs Trust Dubowitz Neuromuscular Centre Long-term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. Journal of Neurology, Neurosurgery & Psychiatry 2012, 84, 698-705, 10.1136/jnnp-2012-303902.
- Sofie Vandevyver; Lien Dejager; Claude Libert; Comprehensive Overview of the Structure and Regulation of the Glucocorticoid Receptor. Endocrine Reviews 2014, 35, 671-693, 10.1210/er.2014-1010.
- Ioanna Petta; Lien Dejager; Marlies Ballegeer; Sam Lievens; Jan Tavernier; Karolien De Bosscher; Claude Libert; The Interactome of the Glucocorticoid Receptor and Its Influence on the Actions of Glucocorticoids in Combatting Inflammatory and Infectious Diseases. Microbiology and Molecular Biology Reviews 2016, 80, 495-522, 10.1128/mmbr.00064-15.
- Robert Newton; Molecular mechanisms of glucocorticoid action: what is important?. Thorax 2000, 55, 603-613, 10.1136/thorax.55.7.603.
- Christopher R. Heier; Jesse M. Damsker; Qing Yu; Blythe C. Dillingham; Tony Huynh; Jack H. Van Der Meulen; Arpana Sali; Brittany K. Miller; Aditi Phadke; Luana Scheffer; et al.James QuinnKathleen TatemSarah JordanSherry DadgarOlga C. RodriguezChris AlbaneseMichael CalhounHeather Gordish-DressmanJyoti K. JaiswalEdward M. ConnorJohn M. McCallEric P. HoffmanErica K. M. ReevesKanneboyina Nagaraju VBP15, a novel anti‐inflammatory and membrane‐stabilizer, improves muscular dystrophy without side effects. EMBO Molecular Medicine 2013, 5, 1569-1585, 10.1002/emmm.201302621.
- Wafaa El-Sankary; Vincent Bombail; G. Gordon Gibson; Nick Plant; Glucocorticoid-mediated induction of CYP3A4 is decreased by disruption of a protein: DNA interaction distinct from the pregnane X receptor response element.. Drug Metabolism and Disposition 2002, 30, 1029-1034, 10.1124/dmd.30.9.1029.
- Michael St. Andre; Mark Johnson; Prashant N. Bansal; Jeremy Wellen; Andrew Robertson; Alan Opsahl; Peter M. Burch; Peter Bialek; Carl A. Morris; Jane Owens; et al. A mouse anti-myostatin antibody increases muscle mass and improves muscle strength and contractility in the mdx mouse model of Duchenne muscular dystrophy and its humanized equivalent, domagrozumab (PF-06252616), increases muscle volume in cynomolgus monkeys. Skeletal Muscle 2017, 7, 25, 10.1186/s13395-017-0141-y.
- Srividya Subramanian; Susan E. Bates; John J. Wright; Igor Espinoza-Delgado; Richard L. Piekarz; Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751-2767, 10.3390/ph3092751.
- Simon Guiraud; Kay E. Davies; Pharmacological advances for treatment in Duchenne muscular dystrophy. Current Opinion in Pharmacology 2017, 34, 36-48, 10.1016/j.coph.2017.04.002.
- Valentina Di Foggia; Xinyu Zhang; Danilo Licastro; Mattia Francesco Maria Gerli; Rahul Phadke; Francesco Muntoni; Philippos Mourikis; Shahragim Tajbakhsh; Matthew Ellis; Laura Greaves†; et al.Robert W. TaylorGiulio CossuLesley G. RobsonSilvia Marino Bmi1 enhances skeletal muscle regeneration through MT1-mediated oxidative stress protection in a mouse model of dystrophinopathy. Journal of Experimental Medicine 2014, 211, 2617-2633, 10.1084/jem.20140317.
- Antonio Filareto; Katie Maguire-Nguyen; Qiang Gan; Garazi Aldanondo; Léo Machado; Jeffrey Chamberlain; Thomas A. Rando; Monitoring disease activity noninvasively in the mdx model of Duchenne muscular dystrophy. Proceedings of the National Academy of Sciences 2018, 115, 7741-7746, 10.1073/pnas.1802425115.
- Sara Nagy; Patricia Hafner; Simone Schmidt; Daniela Rubino-Nacht; Sabine Schädelin; Oliver Bieri; Dirk Fischer; Tamoxifen in Duchenne muscular dystrophy (TAMDMD): study protocol for a multicenter, randomized, placebo-controlled, double-blind phase 3 trial. Trials 2019, 20, 1-14, 10.1186/s13063-019-3740-6.
- Tidhar Turgeman; Yosey Hagai; Kyla Huebner; Judy E. Anderson; Olga Genin; Davinder S Jassal; Arnon Nagler; O Halevy; M. Pines; Prevention of muscle fibrosis and improvement in muscle performance in the mdx mouse by halofuginone Prevention of muscle fibrosis and improvement in muscle performance in the mdx mouse by halofuginone. Neuromuscular Disorders 2008, 18, 857-868, 10.1016/j.nmd.2008.06.386.
- Craig Campbell; Hugh J. McMillan; Jean K. Mah; Mark Tarnopolsky; Kathryn Selby; Ty McClure; Dawn M. Wilson; Matthew L. Sherman; Diana Escolar; Kenneth M. Attie; et al. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial. Muscle & Nerve 2016, 55, 458-464, 10.1002/mus.25268.
- Kyla D. Huebner; Davinder S. Jassal; Orna Halevy; Mark Pines; Judy E. Anderson; Functional resolution of fibrosis in mdx mouse dystrophic heart and skeletal muscle by halofuginone. American Journal of Physiology-Heart and Circulatory Physiology 2008, 294, H1550-H1561, 10.1152/ajpheart.01253.2007.
- Gili Wellner; Sharon Mordechay; Paul Evans; Olga Genin; Mark Pines; Orna Halevy; Hydroxy group requirement for halofuginone-dependent inhibition of muscle fibrosis and improvement of histopathology in the mdx mouse model for Duchenne muscular dystrophy.. Histol. Histopathol. 2019, 34, 791-801.
- Rosemary J Akhurst; Targeting TGF-β Signaling for Therapeutic Gain. Cold Spring Harbor Perspectives in Biology 2017, 9, a022301, 10.1101/cshperspect.a022301.
- Salwa Refat El-Zayat; Hiba Sibaii; Fathia A. Mannaa; Toll-like receptors activation, signaling, and targeting: an overview. Bulletin of the National Research Centre 2019, 43, 1-12, 10.1186/s42269-019-0227-2.
- Wei Zhang; Hui Tu Liu; MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Research 2002, 12, 9-18, 10.1038/sj.cr.7290105.
- Emmanuel Monnet; Ernest H Choy; Iain McInnes; Tamta Kobakhidze; Kathy De Graaf; Philippe Jacqmin; Geneviève Lapeyre; Cristina De Min; Efficacy and safety of NI-0101, an anti-toll-like receptor 4 monoclonal antibody, in patients with rheumatoid arthritis after inadequate response to methotrexate: a phase II study. Annals of the Rheumatic Diseases 2019, 79, 316-323, 10.1136/annrheumdis-2019-216487.
- Iwona Bronisz-Budzyńska; Katarzyna Chwalenia; Olga Mucha; Paulina Podkalicka; Alicja Józkowicz; Agnieszka Łoboda; Magdalena Kozakowska; Jozef Dulak; Karolina- Bukowska- Strakova; miR-146a deficiency does not aggravate muscular dystrophy in mdx mice.. Skeletal Muscle 2019, 9, 22, 10.1186/s13395-019-0207-0.
- Simona Zanotti; Sara Gibertini; Maurizio Curcio; Paolo Savadori; Barbara Pasanisi; Lucia Morandi; Ferdinando Cornelio; Renato Mantegazza; Marina Mora; Opposing roles of miR-21 and miR-29 in the progression of fibrosis in Duchenne muscular dystrophy. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2015, 1852, 1451-1464, 10.1016/j.bbadis.2015.04.013.
- Guoda Ma; Yajun Wang; You Li; Lili Cui; Yujuan Zhao; Bin Zhao; Keshen Li; MiR-206, a Key Modulator of Skeletal Muscle Development and Disease. International Journal of Biological Sciences 2015, 11, 345-352, 10.7150/ijbs.10921.
- G. Berthillier; D. Eichenberger; H.N. Carrier; P. Guibaud; R. Got; Carnitine metabolism in early stages of Duchenne muscular dystrophy. Clinica Chimica Acta 1982, 122, 369-375, 10.1016/0009-8981(82)90140-1.
- Escobar-Cedillo, R.E.; Tintos-Hernandez, J.A.; Martinez-Castro, G.; Montes de Oca-Sanchez, B.; Rodriguez-Jurado, R.; Miranda-Duarte, A.; L-carnitine supplementation in duchenne muscular dystrophy steroid-naive patients: A pilot study. Curr. Top. Nutraceut. Res. 2013, 3, 97–102.
- Erin R. Wissing; Justin G. Boyer; Jennifer Q. Kwong; Michelle A. Sargent; Jason Karch; Elizabeth M. McNally; Kinya Otsu; Jeffery D. Molkentin; P38α MAPK underlies muscular dystrophy and myofiber death through a Bax-dependent mechanism. Human Molecular Genetics 2014, 23, 5452-5463, 10.1093/hmg/ddu270.
- Peter D. Brown; Matrix metalloproteinase inhibitors in the treatment of cancer. Medical Oncology 1997, 14, 1-10, 10.1007/bf02990939.
- A.S. Siegel; S. Henley; A. Zimmerman; M. Miles; R. Plummer; J. Kurz; F. Balch; J.A. Rhodes; G.L. Shinn; C.G. Carlson; et al. The Influence of Passive Stretch and NF-κB Inhibitors on the Morphology of Dystrophic Muscle Fibers. The Anatomical Record: Advances in Integrative Anatomy and Evolutionary Biology 2010, 294, 132-144, 10.1002/ar.21294.
- C. George Carlson; Abbas Samadi; Ashley Siegel; Chronic treatment with agents that stabilize cytosolic IκB-α enhances survival and improves resting membrane potential in MDX muscle fibers subjected to chronic passive stretch. Neurobiology of Disease 2005, 20, 719-730, 10.1016/j.nbd.2005.05.003.
- Ashley L. Siegel; Cathy Bledsoe; Jesse Lavin; Francesca Gatti; Jonas Berge; Gregory Millman; Eric Turin; W. Tyler Winders; John Rutter; Beniamino Palmeiri; et al.C. George Carlson Treatment with inhibitors of the NF-κB pathway improves whole body tension development in the mdx mouse. Neuromuscular Disorders 2009, 19, 131-139, 10.1016/j.nmd.2008.10.006.
- Sonia Messina; Domenica Altavilla; M'hammed Aguennouz; Paolo Seminara; Letteria Minutoli; Maria C. Monici; Alessandra Bitto; Anna Mazzeo; Herbert R. Marini; Francesco Squadrito; et al.Giuseppe Vita Lipid Peroxidation Inhibition Blunts Nuclear Factor-κB Activation, Reduces Skeletal Muscle Degeneration, and Enhances Muscle Function in mdx Mice. The American Journal of Pathology 2006, 168, 918-926, 10.2353/ajpath.2006.050673.
- Nicholas P. Whitehead; Catherine Pham; Othon L. Gervásio; David G. Allen; N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. The Journal of Physiology 2008, 586, 2003-2014, 10.1113/jphysiol.2007.148338.
- Jessica R. Terrill; Hannah G. Radley-Crabb; Miranda D. Grounds; Peter Arthur; N-Acetylcysteine treatment of dystrophic mdx mice results in protein thiol modifications and inhibition of exercise induced myofibre necrosis. Neuromuscular Disorders 2012, 22, 427-434, 10.1016/j.nmd.2011.11.007.
- Rafael De Senzi Moraes Pinto; Renato Ferretti; Luis Henrique Rapucci Moraes; Humberto Santo Neto; Maria Julia Marques; Elaine Minatel; N-Acetylcysteine treatment reduces TNF-α levels and myonecrosis in diaphragm muscle of mdx mice. Clinical Nutrition 2013, 32, 472-475, 10.1016/j.clnu.2012.06.001.
- Gavin Pinniger; Jessica R. Terrill; Evanna B. Assan; Miranda D. Grounds; Peter Arthur; Pre‐clinical evaluation of N‐acetylcysteine reveals side effects in the mdx mouse model of Duchenne muscular dystrophy. The Journal of Physiology 2017, 595, 7093-7107, 10.1113/JP274229.
- Finkel, R.; Vandenborne, K.; Sweeney, H.L.; Finanger, E.; Tennekoon, G.; Shieh, P.; Wilcocks, R.; Walter, G.; Rooney, W.; Forbes, S.C.; et al.et al. Edasalonexent, an NF-κB inhibitor, slows longer-term disease progression on multiple functional and MRI assessments compared to control period in 4 to 7-years old patients with Duchenne muscular dystrophy. Neurology 2019, 92, S51.006.
- Praveen B. Gurpur; Jianming Liu; Dean J Burkin; Stephen J. Kaufman; Valproic Acid Activates the PI3K/Akt/mTOR Pathway in Muscle and Ameliorates Pathology in a Mouse Model of Duchenne Muscular Dystrophy. The American Journal of Pathology 2009, 174, 999-1008, 10.2353/ajpath.2009.080537.
- Annemieke Aartsma-Rus; Ivo F. A. C. Fokkema; Jan J.G.M. Verschuuren; Ieke Ginjaar; Judith Van Deutekom; Gert-Jan Van Ommen; Johan T. Den Dunnen; Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Human Mutation 2009, 30, 293-299, 10.1002/humu.20918.
- Kim M. Keeling; Xiaojiao Xue; Gwen Gunn; D M Bedwell; Therapeutics based on stop codon readthrough.. Annual Review of Genomics and Human Genetics 2014, 15, 371-394, 10.1146/annurev-genom-091212-153527.
- Tatianna Wai Ying Wong; Ronald D. Cohn; Therapeutic Applications of CRISPR/Cas for Duchenne Muscular Dystrophy.. Current Gene Therapy 2017, 17, 301-308, 10.2174/1566523217666171121165046.
- Ryszard Kole; Adrian R. Krainer; Sidney Altman; RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nature Reviews Drug Discovery 2012, 11, 125-140, 10.1038/nrd3625.
- Yu Zhang; Hui Li; Yi-Li Min; Efrain Sanchez-Ortiz; Jian Huang; Alex A. Mireault; John M. Shelton; Jiwoong Kim; Pradeep P. A. Mammen; Rhonda Bassel-Duby; et al.Eric N. Olson Enhanced CRISPR-Cas9 correction of Duchenne muscular dystrophy in mice by a self-complementary AAV delivery system.. Science Advances 2020, 6, eaay6812, 10.1126/sciadv.aay6812.
- Dorota Sienkiewicz; Wojciech Kulak; Bożena Okurowska-Zawada; Grażyna Paszko-Patej; Katarzyna Kawnik; Duchenne muscular dystrophy: current cell therapies. Therapeutic Advances in Neurological Disorders 2015, 8, 166-177, 10.1177/1756285615586123.

