Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Fahim Ahmad | + 2167 word(s) | 2167 | 2021-08-13 09:38:18 | | | |
| 2 | Dean Liu | Meta information modification | 2167 | 2021-08-16 02:41:54 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ahmad, F. Cholesterol Metabolism in Glioblastoma. Encyclopedia. Available online: https://encyclopedia.pub/entry/13162 (accessed on 26 July 2026).
Ahmad F. Cholesterol Metabolism in Glioblastoma. Encyclopedia. Available at: https://encyclopedia.pub/entry/13162. Accessed July 26, 2026.
Ahmad, Fahim. "Cholesterol Metabolism in Glioblastoma" Encyclopedia, https://encyclopedia.pub/entry/13162 (accessed July 26, 2026).
Ahmad, F. (2021, August 13). Cholesterol Metabolism in Glioblastoma. In Encyclopedia. https://encyclopedia.pub/entry/13162
Ahmad, Fahim. "Cholesterol Metabolism in Glioblastoma." Encyclopedia. Web. 13 August, 2021.
Copy Citation
Glioblastoma is a highly lethal adult brain tumor with no effective treatments.
glioblastoma
cholesterol
liver X receptor (LXR)
brain
liver
metabolism
blood–brain barrier
low-density lipoprotein receptor (LDLR)
sterol regulatory element binding protein (SREBP)
1. Pathological and Genetic Features of Glioblastoma
Glioblastoma (also called GBM) is the most common malignant primary brain tumor originating from glial cells [1]. Of the three types of gliomas (ependymomas, oligodendrogliomas, and astrocytomas), glioblastomas are WHO grade IV astrocytomas and have the worst prognosis. GBMs cause 225,000 deaths per year worldwide, and are diagnosed at an average age of 64 [2]. The median survival rate for GBM is 15 months from initial diagnosis even with the most current standard-of-care therapy, which consists of maximal surgical resection, radiation therapy, and adjuvant chemotherapy with temozolomide [3]. There are several obstacles to the development of efficient treatments against glioblastoma. Surgical resection of GBM is virtually impossible as these tumors are highly invasive and penetrate the normal brain. Full resection would require very fine tuned and precise imaging tools, which would enable the removal of invading tumor cells. GBM is highly resistant to cytotoxic drug regimens, including temozolomide, which only improves overall survival 2.5 months beyond radiation and surgery alone [4]. Therefore, new strategies are necessary to develop treatments that elicit durable responses in GBM patients.
Various rodent models have been developed for the accurate representation of preclinical GBM models, but those systems have multiple drawbacks such as immune system deficiency and incompatible stroma and microenvironment that might interfere with the testing new drugs [5]. Existing animal models fall under three categories. The first involves genetically engineered mouse models: for example, mice expressing v-src kinase driven by an astrocyte-specific Glial Fibrillary Acidic Protein (GFAP) promoter or tp53-null mice with astrocyte-specific loss of NF1 both develop high-grade astrocytomas [6][7]. Mouse models of glioblastoma have also been generated using viruses expressing oncogenes injected into the mouse brain. For example, Pax3-Tv-a; Trp53 fl/fl mice injected with RCAS-PDGFB and RCAS-Cre virus, with or without RCAS-H3.3K27M, develop a tumor similar to diffuse pontine glioma [8]. A more recent technical development is the injection of patient-derived glioblastoma stem-like cells in immunocompromised mice. While many laboratories have adopted this technique for studying glioblastoma in vivo, two recent examples include injecting cells derived from isocitrate dehydrogenase 1 (IDH1)-mutant tumors into SCID (Severe Combined Immunodeficiency) mice to study vulnerability of this tumor genotype to 2-hydroxyglutarate depletion [9], and injecting cells from recurrent glioblastoma into NOD-SCID (Non-Obese Diabetic SCID) and NOG (NOD/Shi-scid/IL-2Rγnull) mice then treating them with a STAT3 (Signal Transducer and Activator of Transcription 3) inhibitor [10]. These animal models have led to many novel discoveries in glioma cell biology and metabolism, but thus far have not led to any new clinical advances.
Our most promising avenues for developing robust strategies to target GBM are likely to lie within recent efforts to genomically and proteomically catalog this disease. Indeed, a study published by The Cancer Genome Atlas (TCGA) Research Network in 2014 showed that the most frequently altered pathways in GBM are the RTK/PI3K/MAPK (90% of tumors), p53 (86%), and Rb pathways (79%) [11]. Because nearly all glioblastomas have at least one genomic alteration in the RTK/PI3K/MAPK axis, and because there are numerous small molecule inhibitors targeting this pathway that are already FDA-approved or at least in early-stage clinical trials, it appears inhibition of RTKs, PI3Ks, or MAPKs should be highly effective. Unfortunately, no RTK, PI3K, or MAPK pathway inhibitor thus far has improved patient survival beyond that of the current standard-of-care [4][12][13]. That said, the TCGA datasets have provided a trove of mutations, gene expression and proteomic profiles, and copy number variations that can be explored for novel therapeutic targets.
2. Cholesterol Metabolism in the Liver vs. Brain
The liver plays an important role in cholesterol metabolism. In contrast with brain, hepatic cholesterol can be obtained from the diet. Dietary cholesterol is obtained by intestinal epithelial cells via endocytosis. Cholesterol can then be esterified and loaded into nascent chylomicrons together with triacylglycerol. The chylomicrons are released from intestinal cells into the circulation by the lymphatics [14]. Triacylglycerol in the chylomicrons is hydrolyzed by lipoprotein lipase in blood vessels, and the cholesterol left behind in the chylomicron remnants are taken up and utilized by the liver [14][15]. The cholesterol from liver and dietary origins can be packed into particles of very low-density lipoproteins (VLDLs), which leave the liver and transport cholesterol to other tissues [16]. Another difference between liver and brain cholesterol metabolism is that cholesterol can be recycled through enterohepatic circulation, which does not exist in the brain (Figure 1). Cholesterol can be oxidized in the liver to form bile acids which along with cholesterol is excreted from the liver into the bile [17][18]. The excretion of bile acid involves ABCB11 [19]. Only about 5% of bile acids are lost in the feces, and the rest are reabsorbed into enterocytes. Bile acid is important for the digestion and absorption of dietary fats.
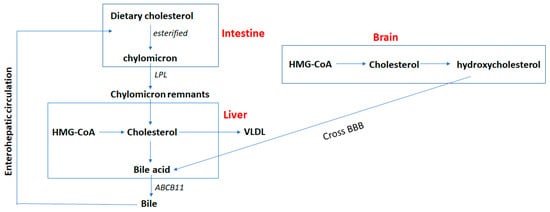
Figure 1. Cholesterol metabolism in liver vs. brain. The brain obtains cholesterol exclusively from de novo synthesis. On the contrary, hepatic cholesterol can be obtained by de novo synthesis and through dietary intake. Dietary cholesterol can be esterified and loaded into chylomicrons in the intestine. The chylomicrons are released into circulation and hydrolyzed by lipoprotein lipase (LPL) to form chylomicron remnants. Cholesterol left behind in the chylomicron remnants are taken up and utilized in the liver. The cholesterol synthesized in liver and from dietary origins can be packed into very low-density lipoprotein (VLDL) and exported from liver. Cholesterol can also be oxidized in the liver to form bile acids which excreted from liver into the bile via the ABCB11 transporter. Cholesterol in the brain can be hydrolyzed to form hydroxycholesterol which crosses the blood–brain barrier (BBB) and goes to the liver to be converted to bile acid. Cholesterol in the liver can be recycled through enterohepatic circulation, which does not exist in the brain. About 5% of the bile acids are lost in the feces, and the rest are reabsorbed into enterocytes.
In summary, cholesterol is involved in cell membrane formation and signaling, and is the precursor of many steroid molecules such as steroid hormones, vitamins, and bile salts. Thus, the metabolism of cholesterol is tightly regulated throughout the body. Although the liver is the primary organ regulating cholesterol homeostasis, the brain cannot uptake cholesterol from peripheral blood and diet due to the BBB (Figure 1). Brain cholesterol is primarily derived from de novo synthesis, and cholesterol levels start to accumulate after birth. Upon reaching adulthood, brain cholesterol levels are maintained at constant levels. Therefore, the excretion of cholesterol from the brain becomes more active in adulthood. Brain cholesterol can be hydroxylated and pass through the BBB to form bile acids in the liver. Moreover, for some types of nerve cells cholesterol must be acquired through the binding of low-density lipoproteins LDLs and LDLRs since cholesterol is mainly synthesized in glial cells and neurons in the adult brain. Disturbed homeostasis of brain cholesterol can lead to diseases such as dementia.
3. Cholesterol Metabolism Pathways Are Altered in Brain Tumors
The brain has different ways to satisfy the requirements of cholesterol compared to peripheral organs. An epidemiological study investigated the relationship between dietary intake of cholesterol and the incidence of cancer, and found that high dietary cholesterol levels increase the risk of several cancers including stomach, colon, rectum, pancreas, lung, breast, kidney, bladder, and non-Hodgkin’s lymphomas, but not brain tumors [20]. This result is not surprising: since cholesterol cannot pass the BBB, high plasma cholesterol levels are unlikely to affect cholesterol metabolism in the brain [20].
The brain obtains cholesterol primarily through de novo synthesis, which involves the mevalonate and Bloch and Kandutsch-Russell pathways [21][22][23]. Taking advantage of the Glioblastoma Bio Discovery Portal [24], our group found a correlation between upregulation of mevalonate and cholesterol pathway and poor survival of GBM patients [25]. Mechanistic studies demonstrated that densely-plated glioma cells increase the synthesis of cholesterol by enhancing oxygen consumption, glycolysis and the pentose phosphate pathway, and pharmacological inhibitors acting downstream of the mevalonate pathway induce glioma cell death [25]. Moreover, the study from our group also found that densely plated normal astrocytes but not tumor sphere glioma cells downregulate genes in the cholesterol biosynthetic pathway including farnesyl diphosphate synthase, farnesyl-diphosphate farnesyltransferase 1, and squalene epoxidase, (FDPS, FDFT1, and SQLE, Figure 2) [25]. A study conducted later by Kim et al. showed that inhibition of FDPS by pharmacological inhibitors and siRNA (small interfering RNA) prevents the formation of secondary spheres of glioma stem cells, and FDPS mRNA was associated with malignancy in glioblastoma patients [26]. In addition to mRNA and protein levels, Abate et al. demonstrate that the activity of FDPS is also upregulated in GBM tumor tissue [27].
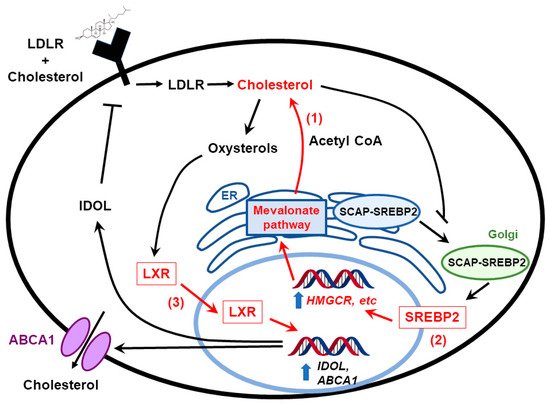
Figure 2. Cholesterol homeostasis in glioblastoma cells. Glioblastoma cells maintain cholesterol under conditions in which normal cells turn it off through multiple mechanisms of dysregulation (highlighted in red). They keep cholesterol biosynthesis on by constitutive activation of the mevalonate pathway (1), and by upregulating SREBPs under hypoxia (2). They are also highly dependent on appropriate levels of LXR activity—hyperactivating LXR with synthetic agonists overstimulates ABCA1 expression and cholesterol efflux, killing glioblastoma (GBM cells) (3). In sum, this provides them with cholesterol in an organ that is blocked from obtaining it from the circulation due to the blood-brain barrier.
Another study demonstrates that GBM is dependent proper cholesterol homeostasis for survival: instead of inhibiting the synthesis of cholesterol, Villa et al. tested the effect of LXRs on treating GBM [28]. LXR is a transcription factor that facilitates the efflux of cholesterol by increasing ABCA1 expression. Villa et al. showed that limiting cholesterol levels by treating cells with LXR agonists induced glioma cell death. In vivo experiments showed that LXR agonists inhibited GBM growth and prolonged the survival of mice [28]. As a key transcription factor in the regulation of sterol homeostasis, other groups have evaluated the effects of SREBPs on GBM development [29][30]. Lewis et al. showed that under hypoxia and serum-deprivation conditions, SREBP is upregulated to maintain the expression of fatty acid and cholesterol biosynthetic genes in GBM cells, and inhibiting SREBP activity under hypoxia led to GBM cell death [29]. These studies demonstrate that cholesterol metabolism pathways are upregulated in GBM patients and targeting cholesterol metabolism and/or homeostasis may be a promising strategy in treating GBM.
4. Targeting Cholesterol Metabolism as a Glioblastoma Therapy
Cancer cells have an increased demand for cholesterol and cholesterol precursors. Therefore, a reasonable assumption is that prevention of tumor-cell growth can be achieved by restricting either cholesterol availability or cholesterol synthesis [31]. Loss of cholesterol feedback inhibition mechanisms that regulate cholesterol synthesis is an important feature of malignant transformation. The cholesterol synthesis pathway has numerous proteins that are potential targets to disrupt cancer progression [32]. The therapeutic potential of targeting these cholesterol synthesis genes is under preclinical investigation [33][34]. The unique metabolic requirements of the brain might make glioblastoma particularly suitable for cholesterol pathway targeting [25].
Liver X-receptors (LXRs) act as transcriptional master switches that coordinate the regulation of genes such as ABCA1 and ABCG1, which are involved in cellular cholesterol homeostasis [35][36] LXR-623 is a synthetic ligand for LXRα and β that upregulates ABCA1 and ABCG1 expression in blood cells [37]. In a study published by the Mischel lab, LXR-623 killed GBM cell lines in an LXR β- and cholesterol-dependent fashion but not healthy brain cells. Upon further investigation of LXR-623, the group found that the drug penetrated the blood–brain barrier and retained its anticancer activity. In addition, mice harboring GBM tumors derived from human patients and treated with LXR-623 had substantially reduced tumor size and improved survival [28].
Statins are HMG-CoA reductase inhibitors that have anti-tumor effects and synergize with certain chemotherapeutic agents to decrease tumor development [34][38]. Several clinical trials have examined the potential chemo-preventive and therapeutic efficacy of different formulations of statins, including simvastatin, pitavastatin, and lovastatin [39][40][41][42][43]. For example, a short-term biomarker study showed lowered breast cancer recurrence in simvastatin-treated patients through the reduction of serum estrone sulfate levels [44]. Moreover, pitavastatin reduces GBM tumor burden in xenografts [40]. Bisphosphonates and tocotrienols are another class of drugs which act as downstream inhibitors of the cholesterol synthesis pathway. Early investigations have shown that they can slow down cancer cell and tumor growth similar to statins [45].
Protein geranylgeranylation, a branch of the cholesterol synthesis pathway, was also found to be essential for maintaining stemness of basal breast cancer cells and to promote human glioma cell growth. GGTI-288, an inhibitor of the geranylgeranyl transferase I (GGTI) reduced the cancer stem cell subpopulation in primary breast cancer xenografts [33][46]. Moreover, high dependency of malignant glioma cells on the isoprenoid pathway for post translational modification of intracellular signaling molecules make this pathway a potential candidate for drug targeting. Recent work published by Ciaglia et al. shows antitumor activity of N6-isopentenyladenosine (iPA) on glioma cells. Its mechanism of action is primarily driven through AMPK-dependent epidermal growth factor receptor (EGFR) degradation, which further adds value to its candidacy as a potent antitumor drug [47][48]. Thus, multiple preclinical studies demonstrate that targeting the cholesterol synthesis pathway could be useful for modulating cancer growth, either through directly inhibiting cholesterol synthesis, or though inhibiting the production of mevalonate pathway-derived moieties used in protein post-translational modifications of oncogenes.
References
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710.
- Alphandery, E. Glioblastoma Treatments: An Account of Recent Industrial Developments. Front. Pharmacol. 2018, 9, 879.
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996.
- Prados, M.D.; Byron, S.A.; Tran, N.L.; Phillips, J.J.; Molinaro, A.M.; Ligon, K.L.; Wen, P.Y.; Kuhn, J.G.; Mellinghoff, I.K.; de Groot, J.F.; et al. Toward precision medicine in glioblastoma: The promise and the challenges. Neuro Oncol. 2015, 17, 1051–1063.
- Miyai, M.; Tomita, H.; Soeda, A.; Yano, H.; Iwama, T.; Hara, A. Current trends in mouse models of glioblastoma. J. Neurooncol. 2017, 135, 423–432. [Green Version]
- Weissenberger, J.; Steinbach, J.P.; Malin, G.; Spada, S.; Rulicke, T.; Aguzzi, A. Development and malignant progression of astrocytomas in GFAP-v-src transgenic mice. Oncogene 1997, 14, 2005–2013. [Green Version]
- Zhu, Y.; Guignard, F.; Zhao, D.; Liu, L.; Burns, D.K.; Mason, R.P.; Messing, A.; Parada, L.F. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell 2005, 8, 119–130. [Green Version]
- Misuraca, K.L.; Hu, G.; Barton, K.L.; Chung, A.; Becher, O.J. A Novel Mouse Model of Diffuse Intrinsic Pontine Glioma Initiated in Pax3-Expressing Cells. Neoplasia 2016, 18, 60–70. [Green Version]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784.
- Ashizawa, T.; Miyata, H.; Iizuka, A.; Komiyama, M.; Oshita, C.; Kume, A.; Nogami, M.; Yagoto, M.; Ito, I.; Oishi, T.; et al. Effect of the STAT3 inhibitor STX-0119 on the proliferation of cancer stem-like cells derived from recurrent glioblastoma. Int. J. Oncol. 2013, 43, 219–227.
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477.
- De Witt Hamer, P.C. Small molecule kinase inhibitors in glioblastoma: A systematic review of clinical studies. Neuro Oncol. 2010, 12, 304–316.
- Thorne, A.H.; Zanca, C.; Furnari, F. Epidermal growth factor receptor targeting and challenges in glioblastoma. Neuro Oncol. 2016, 18, 914–918. [Green Version]
- Dash, S.; Xiao, C.; Morgantini, C.; Lewis, G.F. New Insights into the Regulation of Chylomicron Production. Annu. Rev. Nutr. 2015, 35, 265–294.
- Dietschy, J.M.; Turley, S.D.; Spady, D.K. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J. Lipid Res. 1993, 34, 1637–1659.
- Doonan, L.M.; Fisher, E.A.; Brodsky, J.L. Can modulators of apolipoproteinB biogenesis serve as an alternate target for cholesterol-lowering drugs? Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2018, 1863, 762–771.
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174.
- Midzak, A.; Papadopoulos, V. Binding domain-driven intracellular trafficking of sterols for synthesis of steroid hormones, bile acids and oxysterols. Traffic 2014, 15, 895–914.
- Oude Elferink, R.P.; Groen, A.K. Mechanisms of biliary lipid secretion and their role in lipid homeostasis. Semin. Liver Dis. 2000, 20, 293–305.
- Hu, J.; La Vecchia, C.; de Groh, M.; Negri, E.; Morrison, H.; Mery, L.; Canadian Cancer Registries Epidemiology Research Group. Dietary cholesterol intake and cancer. Ann. Oncol. 2012, 23, 491–500.
- Kandutsch, A.A.; Russell, A.E. Preputial gland tumor sterols. 3. A metabolic pathway from lanosterol to cholesterol. J. Biol. Chem. 1960, 235, 2256–2261.
- Bloch, K. The biological synthesis of cholesterol. Science 1965, 150, 19–28.
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430.
- Celiku, O.; Johnson, S.; Zhao, S.; Camphausen, K.; Shankavaram, U. Visualizing molecular profiles of glioblastoma with GBM-BioDP. PLoS ONE 2014, 9, e101239.
- Kambach, D.M.; Halim, A.S.; Cauer, A.G.; Sun, Q.; Tristan, C.A.; Celiku, O.; Kesarwala, A.H.; Shankavaram, U.; Batchelor, E.; Stommel, J.M. Disabled cell density sensing leads to dysregulated cholesterol synthesis in glioblastoma. Oncotarget 2017, 8, 14860–14875.
- Kim, H.Y.; Kim, D.K.; Bae, S.H.; Gwak, H.; Jeon, J.H.; Kim, J.K.; Lee, B.I.; You, H.J.; Shin, D.H.; Kim, Y.H.; et al. Farnesyl diphosphate synthase is important for the maintenance of glioblastoma stemness. Exp. Mol. Med. 2018, 50, 137.
- Abate, M.; Laezza, C.; Pisanti, S.; Torelli, G.; Seneca, V.; Catapano, G.; Montella, F.; Ranieri, R.; Notarnicola, M.; Gazzerro, P.; et al. Deregulated expression and activity of Farnesyl Diphosphate Synthase (FDPS) in Glioblastoma. Sci. Rep. 2017, 7, 14123. [Green Version]
- Villa, G.R.; Hulce, J.J.; Zanca, C.; Bi, J.; Ikegami, S.; Cahill, G.L.; Gu, Y.; Lum, K.M.; Masui, K.; Yang, H.; et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell 2016, 30, 683–693. [Green Version]
- Lewis, C.A.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D.; et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015, 34, 5128–5140.
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.H.; et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin. Cancer Res. 2016, 22, 5337–5348. [Green Version]
- Buchwald, H. Cholesterol inhibition, cancer, and chemotherapy. Lancet 1992, 339, 1154–1156.
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47.
- Ginestier, C.; Monville, F.; Wicinski, J.; Cabaud, O.; Cervera, N.; Josselin, E.; Finetti, P.; Guille, A.; Larderet, G.; Viens, P.; et al. Mevalonate metabolism regulates Basal breast cancer stem cells and is a potential therapeutic target. Stem Cells 2012, 30, 1327–1337.
- Cruz, P.M.; Mo, H.; McConathy, W.J.; Sabnis, N.; Lacko, A.G. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: A review of scientific findings, relevant to future cancer therapeutics. Front. Pharmacol. 2013, 4, 119.
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Green Version]
- Repa, J.J.; Turley, S.D.; Lobaccaro, J.A.; Medina, J.; Li, L.; Lustig, K.; Shan, B.; Heyman, R.A.; Dietschy, J.M.; Mangelsdorf, D.J. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science 2000, 289, 1524–1529.
- DiBlasio-Smith, E.A.; Arai, M.; Quinet, E.M.; Evans, M.J.; Kornaga, T.; Basso, M.D.; Chen, L.; Feingold, I.; Halpern, A.R.; Liu, Q.Y.; et al. Discovery and implementation of transcriptional biomarkers of synthetic LXR agonists in peripheral blood cells. J. Transl. Med. 2008, 6, 59. [Green Version]
- Warita, K.; Warita, T.; Beckwitt, C.H.; Schurdak, M.E.; Vazquez, A.; Wells, A.; Oltvai, Z.N. Statin-induced mevalonate pathway inhibition attenuates the growth of mesenchymal-like cancer cells that lack functional E-cadherin mediated cell cohesion. Sci. Rep. 2014, 4, 7593. [Green Version]
- Roeder, S.L.; University of Iowa. A Study of the Proper Dosage of Lovastatin and Docetaxel for Patients with Cancer. Available online: https://ClinicalTrials.gov/show/NCT00584012 (accessed on 2 January 2008).
- Claes, A.; Wesseling, P.; Jeuken, J.; Maass, C.; Heerschap, A.; Leenders, W.P. Antiangiogenic compounds interfere with chemotherapy of brain tumors due to vessel normalization. Mol. Cancer Ther. 2008, 7, 71–78. [Green Version]
- Center, L.U.M.; Institute, N.C. Simvastatin and Panitumumab in Treating Patients with Advanced or Metastatic Colorectal Cancer. Available online: https://ClinicalTrials.gov/show/NCT01110785 (accessed on 27 April 2010).
- Center, C.-S.M.; Institute, R.P.C. Cellular Effect of Cholesterol-Lowering Prior to Prostate Removal. Available online: https://ClinicalTrials.gov/show/NCT02534376 (accessed on 27 August 2015).
- Centre, L.H.S. Metformin and Simvastatin Use in Bladder Cancer. Available online: https://ClinicalTrials.gov/show/NCT02360618 (accessed on 10 February 2015).
- Higgins, M.J.; Prowell, T.M.; Blackford, A.L.; Byrne, C.; Khouri, N.F.; Slater, S.A.; Jeter, S.C.; Armstrong, D.K.; Davidson, N.E.; Emens, L.A.; et al. A short-term biomarker modulation study of simvastatin in women at increased risk of a new breast cancer. Breast Cancer Res. Treat. 2012, 131, 915–924.
- Mo, H.; Elson, C.E. Studies of the isoprenoid-mediated inhibition of mevalonate synthesis applied to cancer chemotherapy and chemoprevention. Exp. Biol. Med. 2004, 229, 567–585.
- Zhou, X.; Qian, J.; Hua, L.; Shi, Q.; Liu, Z.; Xu, Y.; Sang, B.; Mo, J.; Yu, R. Geranylgeranyltransferase I promotes human glioma cell growth through Rac1 membrane association and activation. J. Mol. Neurosci. 2013, 49, 130–139.
- Ciaglia, E.; Grimaldi, M.; Abate, M.; Scrima, M.; Rodriquez, M.; Laezza, C.; Ranieri, R.; Pisanti, S.; Ciuffreda, P.; Manera, C.; et al. The isoprenoid derivative N(6)-benzyladenosine CM223 exerts antitumor effects in glioma patient-derived primary cells through the mevalonate pathway. Br. J. Pharmacol. 2017, 174, 2287–2301.
- Ciaglia, E.; Abate, M.; Laezza, C.; Pisanti, S.; Vitale, M.; Seneca, V.; Torelli, G.; Franceschelli, S.; Catapano, G.; Gazzerro, P.; et al. Antiglioma effects of N6-isopentenyladenosine, an endogenous isoprenoid end product, through the downregulation of epidermal growth factor receptor. Int. J. Cancer 2017, 140, 959–972.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
16 Aug 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No