 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jung Gi Kim | + 1324 word(s) | 1324 | 2021-08-10 04:34:07 | | | |
| 2 | Nora Tang | + 649 word(s) | 1973 | 2021-08-13 07:57:47 | | | | |
| 3 | Nora Tang | Meta information modification | 1973 | 2021-08-13 07:58:15 | | |
Video Upload Options
UBR box E3 ligases, also called N-recognins, are integral components of the N-degron pathway. Representative N-recognins include UBR1, UBR2, UBR4, and UBR5, and they bind destabilizing N-terminal residues, termed N-degrons. Understanding the molecular bases of their substrate recognition and the biological impact of the clearance of their substrates on cellular signaling pathways can provide valuable insights into the regulation of these pathways.
1. Introduction
A variety of mechanisms regulate cellular signaling pathways. One such mechanism is the control of protein degradation. Protein degradation serves as a protein homeostasis regulatory network that removes unnecessary proteins from the cellular environment when they are no longer needed, damaged, or misfolded. In eukaryotic cells, the ubiquitin–proteasome system (UPS) and the autophagic–lysosomal pathway are the two major protein degradation systems [1]. Of these, the UPS is responsible for the bulk of intracellular protein degradation (over 80%) and plays an essential regulatory role in critical cellular processes, including cell cycle progression, proliferation, differentiation, angiogenesis, and apoptosis [2][3][4][5][6]. The dysregulation of this pathway is associated with many conditions such as neurodegeneration, cancer, and aging [7][8][9][10][11][12][13].
The UPS utilizes ubiquitin, a 76-amino acid polypeptide, as a tag to mark substrates for degradation. This process is called protein ubiquitination and is mediated by the coordinated action of a cascade of enzymes, including ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), and E3 ubiquitin ligases (E3s) [14][15][16][17]. Protein ubiquitination starts with an E1 enzyme which activates ubiquitin by adenylating its C-terminus. Once activated, ubiquitin is conjugated to an E2 enzyme. Finally, an E3 ubiquitin ligase transfers the ubiquitin from the E2 enzyme to the target protein (substrate). As a result, this process covalently links the C-terminal glycine of ubiquitin to a lysine residue of the target protein through the formation of an isopeptide bond. Thus, E3s are particularly critical players in the ubiquitination process because they determine substrate specificity. There are more than 600 human E3 ubiquitin ligases encoded by approximately 5% of the human genome [18][19][20].
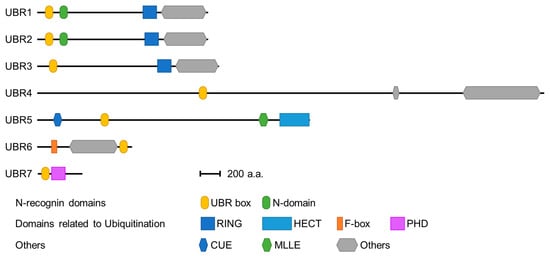
One unique class of E3 ubiquitin ligases (UBR1 to UBR7) is a family that contains an evolutionally conserved UBR box domain, a substrate recognition domain [21][22]. This review discusses the structural features and signaling pathways mediated by these UBR box E3 ligases.
2. N-Degrons and the UBR Box E3 Ligases
According to the N-end rule, the lifespan of a protein depends on the character of its N-terminal residue. N-terminal residues that destabilize a protein are termed N-degrons, classified as type 1 or type 2. Type 1 N-degrons contain positively charged amino acids such as Arg, Lys, and His, and type 2 N-degrons include hydrophobic residues such as Phe, Trp, Tyr, Lue, and Ile. These N-degrons can be generated directly by nonprocessive proteases, including methionine–aminopeptidases (MetAPs), caspases, calpains, separases, or indirectly, by enzymatic cascades that mediate the post-translational arginylation of newly exposed Asn, Gln, Asp, Glu, and Cys in mammals [23][24][25][26][27][28][29][30]. Asn and Gln can be converted to Asp and Glu via deamidation mediated by the protein N-terminal asparagine amidohydrolase (NTAN1) and protein N-terminal glutamine amidohydrolase (NTAQ1), respectively [31][32][33]. N-terminal Cys can be oxidized by oxygen depletion or nitric oxide (NO) to become either Cys-sulfinic acid (CysO 2H) or Cys-sulfonic acid (CysO 3H) [34][35][36]. Recently, the formation of Cys-sulfinic acid has been shown to be mediated by cysteamine (2-aminoethanethiol) dioxygenase (ADO) [37]. N-terminal Asp, Glu, and oxidized Cys are conjugated with the amino acid L-Arg by arginyl-tRNA-protein transferase 1 (ATE1) to generate a canonical N-degron, Arg ( Figure 1 ). Recently, some evidence has shown that stabilizing residues can also act as N-degrons in a context-dependent manner [38].
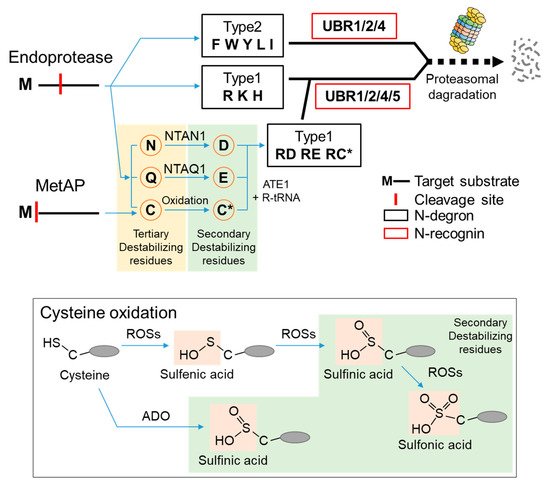
Seven UBR box E3 ligases have been identified in mammals (UBR 1-7) ( Figure 2 ). The UBR box of UBR1, UBR2, UBR4, and UBR5 has been shown to bind type 1 N-degrons. In addition, UBR1 and UBR2 can bind type 2 N-degrons through an N domain present in both proteins [39][40]. However, UBR4 can also bind type 2 N-degrons, although no defined N domain has been identified [39][41]. The molecular mechanism by which UBR4 recognizes type 2 N-degrons requires further investigation. These N-degron-binding UBR box E3 ligases are termed N-recognins.
3. Signaling Pathways Controlled by UBR Box N-Recognins in Mammals
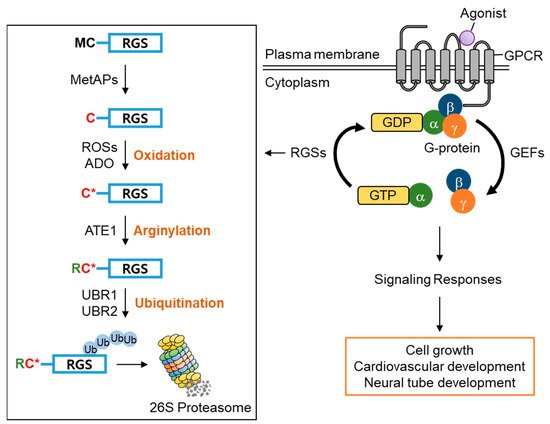
Amongst RGS family proteins, RGS4, RGS5, and RGS16 are known physiological substrates of the Arg/N-degron pathway [34][36][42]. The first methionine residue of RGS4, RGS5, and RGS16 is constantly removed by methionine aminopeptidases (MetAPs), exposing the second cysteine residue, which can be oxidized by reactive oxygen species (ROSs) or cysteamine (2-aminoethanethiol) dioxygenase (ADO) and, in turn, arginylated by arginyltransferase 1 (ATE1) under normoxia [36][37]. The N-terminal Arg of the arginylated RGS proteins is recognized by the UBR box of UBR1/2, which results in poly-ubiquitination and proteasomal degradation. Accordingly, knocking out UBR1/2 has been shown to stabilize RGS4 and RGS5 and exhibited the impairment of neurodevelopment and cardiovascular development in mice, suggesting the importance of UBR1/2′s N-recognin function in controlling G-protein signaling-mediated biological processes [34][36][43][44][45] ( Figure 3 ). Therefore, these RGS proteins have a very transient existence under normoxia due to protein degradation mediated by UBR1/2, prolonging G-protein signaling. However, the stabilization of RGS proteins under hypoxia or the impairment of UBR1/2 restricts G-protein signaling [36].
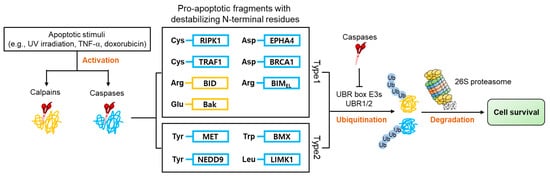
Apoptosis is characterized by the activation of numerous proteases such as caspases and calpains, responsible for the cleavage of over 1000 cellular proteins [46][47][48]. This protease activity generates numerous protein fragments, some of which, termed pro-apoptotic fragments, promote further apoptotic activity in a positive feedback system. Many of these pro-apoptotic fragments have acquired N-degrons ( Figure 4 ) directly from proteolytic cleavage or through the actions of ATE 1, which are recognized by E3 N-recognins and removed via the UPS. Selective removal of pro-apoptotic fragments inhibits apoptosis and promotes cell survival [28][29].
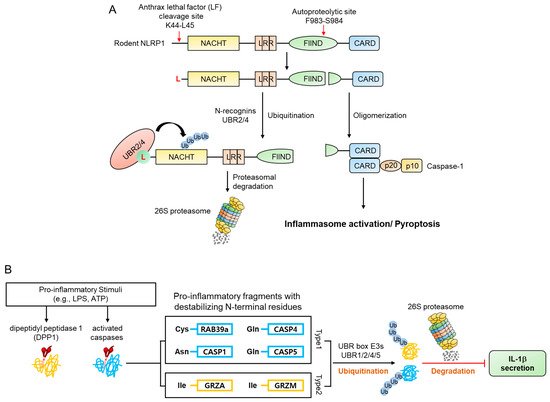
mNLRP1B contains an autocleavage site in the FIIND domain, which consists of the ZU5 and UPA subdomains. N-terminal and C-terminal fragments are generated when mNLRP1B is auto-cleaved between ZU5 and UPA (Phe983-Ser984) [49][50][51]. However, this single cleavage does not result in an inflammatory response due to the autoinhibitory activity of the N-terminal fragment of the protein. Many pathogens utilize mechanisms such as anthrax lethal factor (LF), a metalloprotease and component of anthrax lethal toxin (LT), to target and destroy NLRP proteins in an attempt to evade an immune response. However, when LF enters the cytosol and cleaves mNLRP1B between Lys44 and leu45, it removes the autoinhibitory effect of mNLRP1B and exposes the CARD domain of the C-terminal fragment, inducing pyroptosis and inflammation [49][51][52][53][54] by generating an N-terminal fragment (Leu45-mNLRP1B-F983) containing an N-degron which is recognized by N-recognins such as UBR2 and UBR4 and degraded by the UPS ( Figure 5 A) [55][56]. Accordingly, N-recognins UBR2 and UBR4 were identified through genome-wide CRISPR-Cas9 screening to find proteins related to LT-induced NLRP1 inflammasome activity. Moreover, degradation of the N-terminal fragment of mNLRP1B was significantly reduced due to the deficiency of UBR2 and UBR4 in RAW264.8 cells, and the resistance to LT-induced pyroptosis [55][56].
Replication stress, defined as the slowing or stalling of replication fork progression and DNA synthesis, can cause DNA mutations and chromosomal aberrations [57][58][59]. Thus, failure to counteract genotoxic threats can lead to cancer, developmental disorders, ciliopathies, and laminopathies [58][60][61][62]. Various well-known endogenous and exogenous sources of DNA damage, such as oxidation, chemical mutagens, and ultraviolet radiation, can interfere with the proper progression and completion of the replication process, resulting in genome instability [63][64]. An essential factor of the replication machinery is the proliferating cell nuclear antigen (PCNA) which plays an essential role in maintaining genomic integrity and promoting DNA replication by guiding replicative DNA polymerases at replication forks [65][66]. UV irradiation and various DNA-damaging agents (MMS, mitomycin C, cisplatin, and H 2O 2) lead to the stalling of the replication fork and the release of DNA polymerase from PCNA [64][67][68][69][70][71]. As part of the DNA damage tolerance mechanism, RAD18, a ubiquitin ligase, can monoubiquitinate PCNA, preventing replication fork collapse that can trigger cell death or genome instability by enabling the DNA replication of damaged templates through translesion synthesis (TLS) [72][73]. According to recent studies, SDE2 protein is implicated in genome instability caused by replication stress by modulating this mechanism [74][75].
References
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224.
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797.
- King, B.; Trimarchi, T.; Reavie, L.; Xu, L.; Mullenders, J.; Ntziachristos, P.; Aranda-Orgilles, B.; Perez-Garcia, A.; Shi, J.; Vakoc, C.; et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 2013, 153, 1552–1566.
- Zolk, O.; Schenke, C.; Sarikas, A. The ubiquitin-proteasome system: Focus on the heart. Cardiovasc. Res. 2006, 70, 410–421.
- Rahimi, N. The ubiquitin-proteasome system meets angiogenesis. Mol. Cancer Ther. 2012, 11, 538–548.
- Orlowski, R.Z. The role of the ubiquitin-proteasome pathway in apoptosis. Cell Death Differ. 1999, 6, 303–313.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Tokheim, C.; Wang, X.; Timms, R.T.; Zhang, B.; Mena, E.L.; Wang, B.; Chen, C.; Ge, J.; Chu, J.; Zhang, W.; et al. Systematic characterization of mutations altering protein degradation in human cancers. Mol. Cell 2021, 81, 1292–1308.
- Atkin, G.; Paulson, H. Ubiquitin pathways in neurodegenerative disease. Front. Mol. Neurosci. 2014, 7, 63.
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185.
- Das, C.; Hoang, Q.Q.; Kreinbring, C.A.; Luchansky, S.J.; Meray, R.K.; Ray, S.S.; Lansbury, P.T.; Ringe, D.; Petsko, G.A. Structural basis for conformational plasticity of the Parkinson’s disease-associated ubiquitin hydrolase UCH-L1. Proc. Natl. Acad. Sci. USA 2006, 103, 4675–4680.
- Staub, O.; Gautschi, I.; Ishikawa, T.; Breitschopf, K.; Ciechanover, A.; Schild, L.; Rotin, D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997, 16, 6325–6336.
- Hovsepian, J.; Becuwe, M.; Kleifeld, O.; Glickman, M.H.; Leon, S. Studying Protein Ubiquitylation in Yeast. Methods Mol. Biol. 2016, 1449, 117–142.
- Huang, D.T.; Walden, H.; Duda, D.; Schulman, B.A. Ubiquitin-like protein activation. Oncogene 2004, 23, 1958–1971.
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440.
- Ella, H.; Reiss, Y.; Ravid, T. The Hunt for Degrons of the 26S Proteasome. Biomolecules 2019, 9, 230.
- Garcia-Barcena, C.; Osinalde, N.; Ramirez, J.; Mayor, U. How to Inactivate Human Ubiquitin E3 Ligases by Mutation. Front. Cell Dev. Biol. 2020, 8, 39.
- Medvar, B.; Raghuram, V.; Pisitkun, T.; Sarkar, A.; Knepper, M.A. Comprehensive database of human E3 ubiquitin ligases: Application to aquaporin-2 regulation. Physiol. Genom. 2016, 48, 502–512.
- Yang, B.; Kumar, S. Nedd4 and Nedd4-2: Closely related ubiquitin-protein ligases with distinct physiological functions. Cell Death Differ. 2010, 17, 68–77.
- George, A.J.; Hoffiz, Y.C.; Charles, A.J.; Zhu, Y.; Mabb, A.M. A Comprehensive Atlas of E3 Ubiquitin Ligase Mutations in Neurological Disorders. Front. Genet. 2018, 9, 29.
- Tasaki, T.; Mulder, L.C.; Iwamatsu, A.; Lee, M.J.; Davydov, I.V.; Varshavsky, A.; Muesing, M.; Kwon, Y.T. A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol. Cell. Biol. 2005, 25, 7120–7136.
- Sriram, S.M.; Kim, B.Y.; Kwon, Y.T. The N-end rule pathway: Emerging functions and molecular principles of substrate recognition. Nat. Rev. Mol. Cell Biol. 2011, 12, 735–747.
- Frottin, F.; Martinez, A.; Peynot, P.; Mitra, S.; Holz, R.C.; Giglione, C.; Meinnel, T. The proteomics of N-terminal methionine cleavage. Mol. Cell. Proteom. 2006, 5, 2336–2349.
- Kendall, R.L.; Bradshaw, R.A. Isolation and characterization of the methionine aminopeptidase from porcine liver responsible for the co-translational processing of proteins. J. Biol. Chem. 1992, 267, 20667–20673.
- Rao, H.; Uhlmann, F.; Nasmyth, K.; Varshavsky, A. Degradation of a cohesin subunit by the N-end rule pathway is essential for chromosome stability. Nature 2001, 410, 955–959.
- Varshavsky, A. The N-end rule pathway and regulation by proteolysis. Protein Sci. 2011, 20, 1298–1345.
- Storr, S.J.; Carragher, N.O.; Frame, M.C.; Parr, T.; Martin, S.G. The calpain system and cancer. Nat. Rev. Cancer 2011, 11, 364–374.
- Piatkov, K.I.; Brower, C.S.; Varshavsky, A. The N-end rule pathway counteracts cell death by destroying proapoptotic protein fragments. Proc. Natl. Acad. Sci. USA 2012, 109, E1839–E1847.
- Piatkov, K.I.; Oh, J.H.; Liu, Y.; Varshavsky, A. Calpain-generated natural protein fragments as short-lived substrates of the N-end rule pathway. Proc. Natl. Acad. Sci. USA 2014, 111, E817–E826.
- Xu, Z.; Payoe, R.; Fahlman, R.P. The C-terminal proteolytic fragment of the breast cancer susceptibility type 1 protein (BRCA1) is degraded by the N-end rule pathway. J. Biol. Chem. 2012, 287, 7495–7502.
- Grigoryev, S.; Stewart, A.E.; Kwon, Y.T.; Arfin, S.M.; Bradshaw, R.A.; Jenkins, N.A.; Copeland, N.G.; Varshavsky, A. A mouse amidase specific for N-terminal asparagine. The gene, the enzyme, and their function in the N-end rule pathway. J. Biol. Chem. 1996, 271, 28521–28532.
- Kwon, Y.T.; Balogh, S.A.; Davydov, I.V.; Kashina, A.S.; Yoon, J.K.; Xie, Y.; Gaur, A.; Hyde, L.; Denenberg, V.H.; Varshavsky, A. Altered activity, social behavior, and spatial memory in mice lacking the NTAN1p amidase and the asparagine branch of the N-end rule pathway. Mol. Cell. Biol. 2000, 20, 4135–4148.
- Wang, H.; Piatkov, K.I.; Brower, C.S.; Varshavsky, A. Glutamine-specific N-terminal amidase, a component of the N-end rule pathway. Mol. Cell 2009, 34, 686–695.
- Hu, R.G.; Sheng, J.; Qi, X.; Xu, Z.; Takahashi, T.T.; Varshavsky, A. The N-end rule pathway as a nitric oxide sensor controlling the levels of multiple regulators. Nature 2005, 437, 981–986.
- Kwon, Y.T.; Kashina, A.S.; Davydov, I.V.; Hu, R.G.; An, J.Y.; Seo, J.W.; Du, F.; Varshavsky, A. An essential role of N-terminal arginylation in cardiovascular development. Science 2002, 297, 96–99.
- Lee, M.J.; Tasaki, T.; Moroi, K.; An, J.Y.; Kimura, S.; Davydov, I.V.; Kwon, Y.T. RGS4 and RGS5 are in vivo substrates of the N-end rule pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 15030–15035.
- Masson, N.; Keeley, T.P.; Giuntoli, B.; White, M.D.; Puerta, M.L.; Perata, P.; Hopkinson, R.J.; Flashman, E.; Licausi, F.; Ratcliffe, P.J. Conserved N-terminal cysteine dioxygenases transduce responses to hypoxia in animals and plants. Science 2019, 365, 65–69.
- Varshavsky, A. N-degron and C-degron pathways of protein degradation. Proc. Natl. Acad. Sci. USA 2019, 116, 358–366.
- Tasaki, T.; Zakrzewska, A.; Dudgeon, D.D.; Jiang, Y.; Lazo, J.S.; Kwon, Y.T. The substrate recognition domains of the N-end rule pathway. J. Biol. Chem. 2009, 284, 1884–1895.
- Nillegoda, N.B.; Theodoraki, M.A.; Mandal, A.K.; Mayo, K.J.; Ren, H.Y.; Sultana, R.; Wu, K.; Johnson, J.; Cyr, D.M.; Caplan, A.J. Ubr1 and Ubr2 function in a quality control pathway for degradation of unfolded cytosolic proteins. Mol. Biol. Cell 2010, 21, 2102–2116.
- Tasaki, T.; Kim, S.T.; Zakrzewska, A.; Lee, B.E.; Kang, M.J.; Yoo, Y.D.; Cha-Molstad, H.J.; Hwang, J.; Soung, N.K.; Sung, K.S.; et al. UBR box N-recognin-4 (UBR4), an N-recognin of the N-end rule pathway, and its role in yolk sac vascular development and autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 3800–3805.
- Davydov, I.V.; Varshavsky, A. RGS4 is arginylated and degraded by the N-end rule pathway in vitro. J. Biol. Chem. 2000, 275, 22931–22941.
- An, J.Y.; Seo, J.W.; Tasaki, T.; Lee, M.J.; Varshavsky, A.; Kwon, Y.T. Impaired neurogenesis and cardiovascular development in mice lacking the E3 ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 6212–6217.
- Kim, E.; Kim, S.; Lee, J.H.; Kwon, Y.T.; Lee, M.J. Ablation of Arg-tRNA-protein transferases results in defective neural tube development. BMB. Rep. 2016, 49, 443–448.
- Jiang, Y.; Choi, W.H.; Lee, J.H.; Han, D.H.; Kim, J.H.; Chung, Y.S.; Kim, S.H.; Lee, M.J. A neurostimulant para-chloroamphetamine inhibits the arginylation branch of the N-end rule pathway. Sci. Rep. 2014, 4, 6344.
- Shimbo, K.; Hsu, G.W.; Nguyen, H.; Mahrus, S.; Trinidad, J.C.; Burlingame, A.L.; Wells, J.A. Quantitative profiling of caspase-cleaved substrates reveals different drug-induced and cell-type patterns in apoptosis. Proc. Natl. Acad. Sci. USA 2012, 109, 12432–12437.
- Crawford, E.D.; Seaman, J.E.; Agard, N.; Hsu, G.W.; Julien, O.; Mahrus, S.; Nguyen, H.; Shimbo, K.; Yoshihara, H.A.; Zhuang, M.; et al. The DegraBase: A database of proteolysis in healthy and apoptotic human cells. Mol. Cell Proteom. 2013, 12, 813–824.
- Ono, Y.; Sorimachi, H. Calpains: An elaborate proteolytic system. Biochim. Biophys. Acta 2012, 1824, 224–236.
- Frew, B.C.; Joag, V.R.; Mogridge, J. Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS. Pathog. 2012, 8, e1002659.
- D’Osualdo, A.; Weichenberger, C.X.; Wagner, R.N.; Godzik, A.; Wooley, J.; Reed, J.C. CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5-like domain. PLoS ONE 2011, 6, e27396.
- Finger, J.N.; Lich, J.D.; Dare, L.C.; Cook, M.N.; Brown, K.K.; Duraiswami, C.; Bertin, J.; Gough, P.J. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J. Biol. Chem. 2012, 287, 25030–25037.
- Levinsohn, J.L.; Newman, Z.L.; Hellmich, K.A.; Fattah, R.; Getz, M.A.; Liu, S.; Sastalla, I.; Leppla, S.H.; Moayeri, M. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 2012, 8, e1002638.
- Hellmich, K.A.; Levinsohn, J.L.; Fattah, R.; Newman, Z.L.; Maier, N.; Sastalla, I.; Liu, S.; Leppla, S.H.; Moayeri, M. Anthrax lethal factor cleaves mouse nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PLoS ONE 2012, 7, e49741.
- Chavarria-Smith, J.; Vance, R.E. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog. 2013, 9, e1003452.
- Chui, A.J.; Okondo, M.C.; Rao, S.D.; Gai, K.; Griswold, A.R.; Johnson, D.C.; Ball, D.P.; Taabazuing, C.Y.; Orth, E.L.; Vittimberga, B.A.; et al. N-terminal degradation activates the NLRP1B inflammasome. Science 2019, 364, 82–85.
- Xu, H.; Shi, J.; Gao, H.; Liu, Y.; Yang, Z.; Shao, F.; Dong, N. The N-end rule ubiquitin ligase UBR2 mediates NLRP1B inflammasome activation by anthrax lethal toxin. EMBO. J. 2019, 38, e101996.
- Wilhelm, T.; Said, M.; Naim, V. DNA Replication Stress and Chromosomal Instability: Dangerous Liaisons. Genes 2020, 11, 642.
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9.
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109.
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289.
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543.
- Broers, J.L.; Hutchison, C.J.; Ramaekers, F.C. Laminopathies. J. Pathol. 2004, 204, 478–488.
- Friedberg, E.C. A brief history of the DNA repair field. Cell Res. 2008, 18, 3–7.
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204.
- Essers, J.; Theil, A.F.; Baldeyron, C.; van Cappellen, W.A.; Houtsmuller, A.B.; Kanaar, R.; Vermeulen, W. Nuclear dynamics of PCNA in DNA replication and repair. Mol. Cell Biol. 2005, 25, 9350–9359.
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679.
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500.
- Lehmann, A.R.; Niimi, A.; Ogi, T.; Brown, S.; Sabbioneda, S.; Wing, J.F.; Kannouche, P.L.; Green, C.M. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair. 2007, 6, 891–899.
- Li, X.Q.; Ren, J.; Chen, P.; Chen, Y.J.; Wu, M.; Wu, Y.; Chen, K.; Li, J. Co-inhibition of Pol eta and ATR sensitizes cisplatin-resistant non-small cell lung cancer cells to cisplatin by impeding DNA damage repair. Acta Pharmacol. Sin. 2018, 39, 1359–1372.
- Kashiwaba, S.; Kanao, R.; Masuda, Y.; Kusumoto-Matsuo, R.; Hanaoka, F.; Masutani, C. USP7 Is a Suppressor of PCNA Ubiquitination and Oxidative-Stress-Induced Mutagenesis in Human Cells. Cell Rep. 2015, 13, 2072–2080.
- Niimi, A.; Brown, S.; Sabbioneda, S.; Kannouche, P.L.; Scott, A.; Yasui, A.; Green, C.M.; Lehmann, A.R. Regulation of proliferating cell nuclear antigen ubiquitination in mammalian cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16125–16130.
- Hibbert, R.G.; Huang, A.; Boelens, R.; Sixma, T.K. E3 ligase Rad18 promotes monoubiquitination rather than ubiquitin chain formation by E2 enzyme Rad6. Proc. Natl. Acad. Sci. USA 2011, 108, 5590–5595.
- Zhang, W.; Qin, Z.; Zhang, X.; Xiao, W. Roles of sequential ubiquitination of PCNA in DNA-damage tolerance. FEBS. Lett. 2011, 585, 2786–2794.
- Jo, U.; Cai, W.; Wang, J.; Kwon, Y.; D’Andrea, A.D.; Kim, H. PCNA-Dependent Cleavage and Degradation of SDE2 Regulates Response to Replication Stress. PLoS Genet. 2016, 12, e1006465.
- Rageul, J.; Park, J.J.; Jo, U.; Weinheimer, A.S.; Vu, T.T.M.; Kim, H. Conditional degradation of SDE2 by the Arg/N-End rule pathway regulates stress response at replication forks. Nucleic Acids Res. 2019, 47, 3996–4010.

