Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | NORHAIZAN MOHD ESA | + 2894 word(s) | 2894 | 2021-08-05 05:17:27 | | | |
| 2 | Peter Tang | Meta information modification | 2894 | 2021-08-13 03:11:57 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mohd Esa, N. High-Fat Diets-Mediated Oxidative Stress. Encyclopedia. Available online: https://encyclopedia.pub/entry/13126 (accessed on 28 June 2026).
Mohd Esa N. High-Fat Diets-Mediated Oxidative Stress. Encyclopedia. Available at: https://encyclopedia.pub/entry/13126. Accessed June 28, 2026.
Mohd Esa, Norhaizan. "High-Fat Diets-Mediated Oxidative Stress" Encyclopedia, https://encyclopedia.pub/entry/13126 (accessed June 28, 2026).
Mohd Esa, N. (2021, August 13). High-Fat Diets-Mediated Oxidative Stress. In Encyclopedia. https://encyclopedia.pub/entry/13126
Mohd Esa, Norhaizan. "High-Fat Diets-Mediated Oxidative Stress." Encyclopedia. Web. 13 August, 2021.
Copy Citation
Cognitive dysfunction is linked to chronic low-grade inflammatory stress that contributes to cell-mediated immunity in creating an oxidative environment. Food is a vitally important energy source; it affects brain function and provides direct energy. Several studies have indicated that high-fat consumption causes overproduction of circulating free fatty acids and systemic inflammation. Immune cells, free fatty acids, and circulating cytokines reach the hypothalamus and initiate local inflammation through processes such as microglial proliferation.
cognitive impairment
high-fat diet
inflammation
neurodegeneration
oxidative stress
1. Introduction
The prevalence of obesity worldwide has doubled from 1980 to 2008 [1]. Individuals who are overweight or obese are at risk of developing metabolic syndrome (hyperlipidemia, type 2 diabetes, hypertension, and hypercholesterolemia) [2] and psychiatric conditions, for instance depression [3] and anxiety disorders [4]. Dietary intake of fat has markedly increased from 1991 to 2008 [5]. Substantial evidence highlights the detrimental impact of diets high in saturated fat [6], and extensive research has shown that diet-induced obesity potentially leads to memory impairment in rodents [7]. Mice fed a long-term high-fat diet (HFD) show reduced learning and memory performance [8], as well as depressive- [9][10] and anxiety-like behaviors [11][12].
Oxidative stress plays a crucial role in the development of numerous diseases [13]. Reactive nitrogen species (RNS) and reactive oxygen species (ROS) are continuously produced in the body via mitochondrial bioenergetics and oxidative metabolism [14]. Compelling evidence reveals that overproduction of ROS causes cumulative oxidative damage to macromolecules, including DNA, proteins, and membrane lipids [15], leading to neuronal death and affecting the healthspan of several organ systems [16]. High consumption of dietary fat contributes to obesity, which induces a permanent state of inflammation via the generation of white adipose tissue that secretes proinflammatory factors [17]. A previous study demonstrated that activated immune cells produce high levels of ROS, primarily mediated by nuclear factor kappa B (NF-κB) and proinflammatory cytokines [18].
Oxidative stress plays a prominent role in the development of neurodegenerative diseases [19]. The brain is highly sensitive and vulnerable to oxidation due to the presence of large amounts of unsaturated fatty acids and oxygen that serve as a substrate for lipid peroxidation [20]. Peroxidation products of fatty acids are among the biomarkers of oxidative stress in neurodegenerative diseases, along with protein nitration and carbonylation, and RNA and DNA oxidative damages [21]. Neurodegenerative diseases are often linked to abnormal protein aggregation. Such abnormal protein aggregation is able to induce oxidative stress through ROS production and mitochondria dysfunction [22][23]. Food is a vitally important energy source; it provides direct energy and affects brain function. Therefore, the role of high-fat diet (HFD) modulating neurodegeneration and oxidative stress is worthy of further discussion.
2. HFD-Mediated Oxidative Stress
Diets high in nutrient-dense foods, monounsaturated fatty acids (MUFAs), and dietary fibers have recently been replaced by diets high in saturated fats and refined sugars [24]. Fatty acids interact with various transcriptional factors to trigger downstream signaling pathways [25]. Among all transcription molecules, peroxisome proliferator-activated receptor (PPAR) is a ligand-activated transcription factor that acts as a sensor for lipid-regulating proteins [26]. Polyunsaturated fatty acids (PUFAs) interact with sterol regulatory element binding proteins (SREBP) and transcription factors in the liver may modulate genes associated with their synthesis and facilitate phospholipid, fatty acid, and cholesterol uptake [27]. Studies have revealed a positive association between body adiposity and HFD (r = 0.57, p = 0.0002), particularly saturated fat [28]. High body adiposity may induce the production of ROS, accompanied by elevated adipokine and tumor necrosis factor alpha (TNF-α) secretion, which promotes chronic inflammation [29].
Fatty acids are involved in catabolism through peroxisomal β-oxidative and mitochondrial pathways [30]. Mitochondrial β-oxidation is primarily responsible for the degradation of long, medium, and short chain fatty acids (≤18 C), whereas peroxisomal β-oxidation participates in the catabolism of branch chain fatty acids, unsaturated fatty acids, dicarboxylic acids, and very long chain fatty acids (≥20 C) [31][32]. Additionally, peroxisomal β-oxidation is also involved in the synthesis of certain fatty acids, for instance, docosahexaenoic acid (DHA) [33]. Abnormalities in the peroxisomal β-oxidation pathway have been characterized by an accumulation of very long chain fatty acids and progressive neurological dysfunction, including dementia [31]. In particular, PPARα plays a crucial role in regulating fatty acid β-oxidation gene expression for factors such as catalase, acyl-CoA oxidase 1 (ACOX1), and carnitine-palmitoyl transferase-I (CPT-I) [34].
Indeed, the liver is the predominant organ for fatty acid oxidation in the body [35]. A previous study demonstrated that peroxisomal function was inversely correlated with aging [36]. Reduced ACOX1 expression and peroxisomal fatty acid β-oxidation activity have been observed in the liver of old rats [37]. In another study, Sanguino et al. [38] revealed that aging strongly decreases the activity and expression of PPARα in rat liver. These findings imply that fatty acid β-oxidation activity is reduced with age, subsequently resulting in reduced levels of hepatic PPARα [39].
In addition, several studies reported by O’Brien et al. [40] and Okereke et al. [41] have shown an association (p for linear trend = 0.008, odds ratio (OR) with 95% confidence interval (CI) = 1.64) between excessive fat consumption and neurological dysfunction. Data indicated that excessive fat consumption results in a net energy overload, which in turn leads to the expansion of adipose tissue. Sustained chronic fat intake may contribute to adipose tissue dysfunction and metabolic inflammation. When circulating free fatty acids increase, they can induce lipotoxicity to peripheral tissues, such as liver and β-cell dysfunction [40]. The high influx of free fatty acids into the liver triggers very low-density lipoprotein (VLDL) triglycerides production and contributes to the development of dyslipidemia. Subsequently, these impairments can lead to metabolic syndromes. The central nervous system is also adversely affected by elevated levels of circulating free fatty acids and the lipotoxic effects of dyslipidemia [40]. Ultimately, these dysfunctions contribute to neurological disorders, such as mild cognitive impairment and Alzheimer’s disease [40].
Furthermore, HFD increases levels of chylomicrons in the intestine. These chylomicrons enter circulation and cause the generation of free fatty acids, which are taken up by the liver. These hepatic free fatty acids may either enter the mitochondria for β-oxidation or be esterified into triglycerides. Triglycerides either accumulate in hepatocytes as small droplets or produce VLDL, which is then converted into low-density lipoprotein (LDL) [42]. Excessive LDL burden in the blood may form oxidized-LDL (Ox-LDL) due to its excessive accumulation or lack of LDL-receptors in hepatocytes, which in turn is engulfed by macrophages and becomes foam cells. Subsequently, foam cells accumulate in the arterial endothelium to form plaques. Ultimately, these plaques lead to cardiovascular and circulatory disorders and increase blood-brain barrier permeability [43].
Additionally, mitochondrial β-oxidation of free fatty acids is linked to the conversion of oxidized cofactors (FAD and NAD+) into reduced cofactors, FADH2 and NADH and is thereby reoxidized and restored back into FAD and NAD+ by the mitochondrial respiratory chain. During reoxidation, NADH and FADH2 transfer electrons to the first complexes of the respiratory chain. These electrons then migrate up to cytochrome c oxidase and combine with oxygen and protons to form water. These intermediates may interact with oxygen to produce increasing levels of ROS and superoxide anion radicals [44][45]. Therefore, overconsumption of fat triggers mitochondrial β-oxidation of free fatty acids, subsequently leading to excess electron flow using cytochrome c oxidase, which increases ROS accumulation. Mitochondria are a crucial cellular source of ROS; they oxidize unsaturated lipids of fat deposits to cause lipid peroxidation. HFD-induced ROS may trigger proinflammatory signaling and activate NF-κB transcriptional factor, and thus inducing NF-κB-dependent proinflammatory molecules, such as interferon-γ (IFN-γ), TNF-α, and inducible nitric oxide synthase (iNOS) [46]. In addition to ROS production, overproduction of nitric oxide (NO) via the activation of iNOS also causes accumulation of RNS [47].
3. Association of a High-Fat Diet with Cognitive Function
3.1. Animal Study
A previous study has demonstrated that rodents fed a diet high in saturated fatty acids (SFAs) show increased brain inflammatory markers [48]. Increased body weight and adiposity can cause neurological perturbations. These data imply that excess adipose tissue is highly metabolically active and susceptible to the release of proinflammatory mediators [49]. Further, data from a previous study also demonstrated that triglyceride administration impairs hippocampal long-term potentiation [50]. Interestingly, the mice treated with HFD for only one day is sufficient to induce a rapid drop in the performance of episodic memory task [51]. This study further revealed that memory deficits are rapidly reversed when switching mice to a low-fat diet from a HFD [51]. In another study, Duffy et al. [52] found that orexin/ataxin-3 (O/A3) mice, a transgenic mouse model of orexin neurodegeneration, fed with HFD accelerate two-way active avoidance (TWAA) hippocampus-dependent memory task and the onset of neuroinflammation. In a study by Woodie and Blythe [53] focusing on hypercaloric diet (high in fat and fructose) and its effects on cognitive health, the rats fed with a HFD exhibited a relatively high fat pad weight compared to the high-fructose group. This study further demonstrated that high-fructose diet promotes insulin dysregulation, development of hyperlipidemia, and impairs cognitive performance. However, no apparent cognitive deficits were observed in rats fed with a HFD [53]. These observations indicate that an individual part of the hypercaloric diet may cause negative impacts on metabolic syndrome and cognitive function, suggesting the combination of high-fructose and high-fat components may further aggravate physiological issues and be detrimental to human health. In support of this, the animal study has shown that hypercaloric diets altered lipid and energy metabolism similar to clinical diabetes, with elevation of fasting glucose and increased cholesterol levels. This study further revealed that hypercaloric diets can impair spatial learning ability and synaptic plasticity [54]. These adverse effects were also found in rats fed with a HFD, suggesting that insulin resistance is a probable mediator to HFD-induced cognitive deficits [55].
3.2. Human Study
A recent study stated that Western-style diets, which are high in refined sugars and saturated fats, have a greater likelihood of inducing memory impairment in healthy subjects [56]. Data from human studies investigating the impact of HFD on cognitive function are limited, but evidence from an intervention study indicates an association between HFD consumption and cognitive disorder. Holloway et al. [57] found that HFD administration (nearly 75% of energy) for five days is sufficient to induce depression and impair retrieval speed and attention. Consistent with a study reported by Holloway et al. [57], Edwards et al. [58] demonstrated that HFD consumption for seven days reduced attention and reaction time in sedentary adult males with fewer than 2 h/week physical activity. Similarly, HFD was also found to induce neuroinflammation. A study reported by Mittal and Katare [59] also demonstrated that high triglyceride levels convey poor cognitive performance in type 2 diabetes patients. Moreover, a study by Okereke et al. [41] also found that increased intake of saturated fats in young adults impairs prospective memory, cognitive function, and memory speed and flexibility, leading to neurological diseases, such as dementia and Alzheimer’s disease in mid and later life. Although many studies have demonstrated that HFD increased the risk of cognitive disorders; not all data demonstrated such a link [60]. Several studies reported by Solfrizzi et al. [61], Cherbuin and Anstey [62], and Roberts et al. [63] did not identify an association between high SFA intake and mild cognitive impairment. Variability in study designs may partly explain the inconsistency in findings. For instance, the variation in means of reporting composition and type of dietary fats and its association with cognitive problems shows a barrier in combining the findings of studies. The investigators used different methods for defining fat types (for example, PUFA from spreads or total PUFA) may address different subgroups (for instance, those with diabetes). Furthermore, the small sample size may increase the likelihood of inconsistent findings between the studies. In fact, the small sample size of study makes it difficult to detect significant associations. The nonsignificant findings could be attributed to the lack of statistical power. In addition, by comparing different study groups with diverse populations and its relationship with cognition may also explain inconsistency in findings. Individuals from different human populations have different proportions of genetic. Genetic variation in a population is derived from a wide assortment of alleles and genes. The persistence of certain populations over time via changing environments is highly dependent on their ability to shift or adapt external conditions.
In addition, data from a Rotterdam cohort study involving 197 cases of dementia, including 29 with vascular dementia, 146 with Alzheimer’s disease, and 22 with other types failed to show adverse outcome of high SFA intake and dementia risk after 6 years of follow-up [64]. Furthermore, data from Washington Heights-Inwood Columbia Aging Project (WHICAP) involving 980 New Yorkers aged 65 years and above, in which 28% of them carried APOE ε4 allele, showed that high SFA intake was not associated with the increased risk of Alzheimer’s disease [65]. Specifically, data demonstrate that compared to an omega-6 PUFA diet, Alzheimer’s disease risk was reduced in subjects consuming an omega-3 PUFA diet [66][67]. In contrast, high omega-3 PUFA intake is not associated with long-term dementia risk [68] (p for linear trend = 0.7, hazard ratio (HR) = 0.95; 95% CI = 0.76, 1.19). Taken together, these findings demonstrate an association between HFD and cognitive loss, suggesting potentially causal roles of HFD and brain inflammation in driving cognitive disruption. Figure 1 shows the effects of HFD on cognitive function.
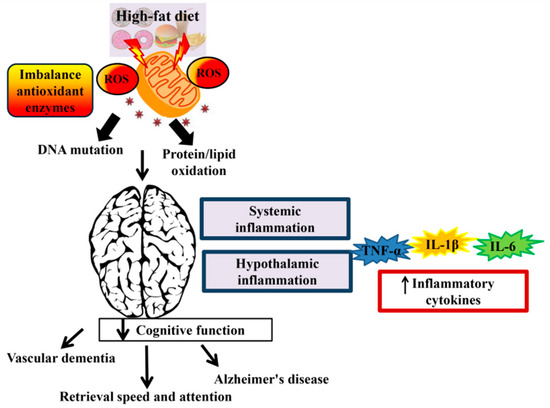
Figure 1. The effect of a high-fat diet (HFD) on cognitive function. Consumption of HFD induces reactive oxygen species (ROS). Accumulation of ROS leads to DNA mutation and protein/lipid oxidation and subsequently reduced the mitochondrial function. Overproduction of reactive species that occur in the mitochondrial DNA can lead to neurodegenerative disease and brain dysfunction. Systemic inflammation and hypothalamic inflammation promotes cognitive decline via secretion of inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6).
The studies had to meet the following criteria: exposure to HFD (PUFA, total, saturated, trans fat, or cholesterol) in rodents (rats or mice) or at any adult age in human; endpoints included cognitive function, incident mild cognitive impairment, Alzheimer’s disease, or dementia; or cognitive decline; in studies assessing dietary assessment and determination of cognitive function or outcome (mild cognitive impairment, Alzheimer’s disease, or dementia) or in studies assessing cognitive decline. There was no restriction pertaining to the publication date, sample size or the number of animals, animal strains, ethnicity of participants or on language, race, or gender. Exclusions criteria in animal study should also be considered. First, HFD was not administered. Second, other types of animals (dogs, cats, or sheep) were used. Third, duplicate publications. Moreover, case reports and studies limited to individuals with medical conditions (dyslipidemia or CVD) that are likely to affect intervention trials and cognitive status were excluded because of the confounding or bias in their study designs.
Data from the animal experiments have shown a greater likelihood of inflammation after the administration of HFD. Importantly, some research has emerged to suggest the detrimental impact of HFD on gene expression in hypothalamus [69][70][71]. In particular, a HFD led to an upregulation of genes such as toll-like receptor 4 (TLR4), NF-κB, Cd68, Emr1, IL-6, indoleamine 2,3-dioxygenase (IDO), TNF-α, and interferon gamma (IFN-γ) in rodents, suggesting that these changes were related to diet-induced changes rather than obesity. In line with this, human studies also showed an association between HFD and Alzheimer’s disease, mild cognitive impairment, or dementia. These relationships could be partly due to the type or composition of dietary fatty acids that may influence the cognitive function. In fact, the methodological quality of human studies may fail to assess precision outcomes. Most of the human studies using semiquantitative food-frequency questionnaire (FFQ) to measure the habitual intake during the study period and do not account for long-term periods or for day-to-day variation of intake. The measures obtained from the FFQ may not have enough precision to make inferences on the absolute amounts of nutrient intake that linked to the occurrence of cognitive outcomes. Although several limitations and potential publication bias may undermine the validity of the findings, HFD may play a potential role in neurodegenerative disease. Overall, these data imply that HFD consumption, particularly SFA may serve as a stimulus to elevate the inflammatory markers and augmented the inflammatory response, and ultimately lead to brain dysfunction and neurodegenerative disease.
4. Mechanisms Responsible for High-Fat Diet-Induced Cognitive Deficits
Substantial evidence suggests that increased oxidative stress and altered apoptosis contribute to the pathogenesis of neurodegenerative diseases [72]. Free radicals are implicated in the progression and development of cognitive deficits by interrupting synaptic transmission, mitochondrial function, neuroinflammation, and axonal transport; all of these factors contribute to neuronal loss in Alzheimer’s and other dementia diseases [73]. In fact, mitochondria are involved in the pathogenesis of numerous neurodegenerative diseases. Mitochondria are the predominant organelles that supply ATP to cells via oxidative phosphorylation, respond to oxidative stress, and synthesize additional key molecules. Mitochondria produce redox enzymes that are required for transferring electrons from one substrate to another, and inefficiencies in this process may lead to ROS production. The central nervous system is highly dependent on mitochondrial function due to its high energy demands. Mutation and ROS production that occurs in the mitochondrial DNA can lead to neurodegenerative disease and brain dysfunction. Indeed, many neurodegenerative diseases have been linked to mitochondrial dysfunction [72].
Accumulation of mitochondria DNA mutations during aging may cause malfunctioning oxidative phosphorylation and an imbalance in antioxidant enzymes. Mitochondrial dysfunction produces the ROS axis and forms a vicious cycle, which is regarded as the basis of the mitochondrial free radical theory of aging. A previous study revealed that ROS plays a critical role in the regulation of several cellular metabolic processes, including antioxidant defense mechanisms and posttranslational modification of proteins. However, oxidative stress in normal homeostasis is disturbed during aging and subsequently increases intracellular ROS levels [74]. Production of oxidative stress affects the mitochondrial defense system and membrane permeability, influences Ca2+ homeostasis, deranges the mitochondrial respiratory chain, and causes DNA mutations in mitochondria. This phenomenon triggers neurodegeneration and amplifies neuronal dysfunction [75].
References
- World Health Organization. Obesity and Overweight. 2015. Available online: http://www.who.int/mediacentre/factsheets/fs311/en/2015 (accessed on 26 November 2018).
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2017, 114, 1752–1761.
- Milaneschi, Y.; Lamers, F.; Bot, M.; Drent, M.L.; Penninx, B.W.J.H. Leptin Dysregulation is specifically associated with major depression with atypical features: Evidence for a mechanism connecting obesity and depression. Biol. Psychiatry 2017, 81, 807–814.
- De Noronha, S.R.; Campos, G.V.; Abreu, A.R.; de Souza, A.A.; Chianca, D.A.; de Menezes, R.C. High fat diet induced-obesity facilitates anxiety-like behaviors due to GABAergic impairment within the dorsomedial hypothalamus in rats. Behav. Brain Res. 2017, 316, 38–46.
- Vadiveloo, M.; Scott, M.; Quatromoni, P.; Jacques, P.; Parekh, N. Trends in dietary fat and high-fat food intakes from 1991 to 2008 in the Framingham heart study participants. Br. J. Nutr. 2014, 111, 724–734.
- DiNicolantonio, C.J.; Lucan, S.C.; O’Keefe, J.H. The evidence for saturated fat and sugar related to coronary heart disease. Prog. Cardiovasc. Dis. 2016, 58, 464–472.
- Alzoubi, K.H.; Mayyas, F.A.; Mahafzah, R.; Khabour, O.F. Melatonin prevents memory impairment induced by high-fat diet: Role of oxidative stress. Behav. Brain Res. 2018, 336, 93–98.
- Cordner, Z.A.; Tamashiro, K.L. Effects of high-fat diet exposure on learning and memory. Physiol. Behav. 2015, 152, 363–371.
- Kurhe, Y.; Mahesh, R.; Gupta, D. Effect of a selective cyclooxygenase type 2 inhibitor celecoxib on depression associated with obesity in mice: An approach using behavioral tests. Neurochem. Res. 2014, 39, 1395–1402.
- Kurhe, Y.; Mahesh, R. Ondansetron attenuates co-morbid depression and anxiety associated with obesity by inhibiting the biochemical alterations and improving serotonergic neurotransmission. Pharm. Biochem. Behav. 2015, 136, 107–116.
- Sivanathan, S.; Thavartnam, K.; Arif, S.; Elegino, T.; McGowan, P.O. Chronic high fat feeding increases anxiety-like behaviour and reduces transcript abundance of glucocorticoid signaling genes in the hippocampus of female rats. Behav. Brain Res. 2015, 286, 265–270.
- Zemdegs, J.; Quesseveur, G.; Jarriault, D.; Pénicaud, L.; Fioramonti, X.; Guiard, B.P. High-fat diet-induced metabolic disorders impairs 5-HT function and anxiety-like behavior in mice. Br. J. Pharm. 2016, 173, 2095–2110.
- Tan, B.L.; Norhaizan, M.E.; Liew, W.-P.-P. Nutrients and oxidative stress: Friend or foe? Oxid. Med. Cell. Longev. 2018, 2018.
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619.
- Wang, L.; Chen, X.; Du, Z.; Li, G.; Chen, M.; Chen, X.; Liang, G.; Chen, T. Curcumin suppresses gastric tumor cell growth via ROS-mediated DNA polymerase γ depletion disrupting cellular bioenergetics. J. Exp. Clin. Cancer Res. 2017, 36, 47.
- Mazon, J.N.; de Mello, A.H.; Ferreira, G.K.; Rezin, G.T. The impact of obesity on neurodegenerative diseases. Life Sci. 2017, 182, 22–28.
- Muñoz, A.; Costa, M. Nutritionally mediated oxidative stress and inflammation. Oxidat. Med. Cell. Longev. 2013, 2013.
- Knight, J.A. Review: Free radicals, antioxidants, and the immune system. Ann. Clin. Lab. Sci. 2000, 30, 145–158.
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. Febs Lett. 2018, 592, 692–702.
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503.
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic. Biol. Med. 2007, 43, 658–677.
- Selfridge, J.E.; Lezi, E.; Lu, J.; Swerdlow, R.H. Role of mitochondrial homeostasis and dynamics in Alzheimer’s disease. Neurobiol. Dis. 2013, 51, 3–12.
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 316523.
- Marventano, S.; Godos, J.; Platania, A.; Galvano, F.; Mistretta, A.; Grosso, G. Mediterranean diet adherence in the Mediterranean healthy eating, aging and lifestyle (MEAL) study cohort. Int. J. Food Sci. Nutr. 2018, 69, 100–107.
- Du, X.; Zhu, Y.; Peng, Z.; Cui, Y.; Zhang, Q.; Shi, Z.; Guan, Y.; Sha, X.; Shen, T.; Yang, Y.; et al. High concentrations of fatty acids and β-hydroxybutyrate impair the growth hormone-mediated hepatic JAK2-STAT5 pathway in clinically ketotic cows. J. Dairy Sci. 2018, 101, 3476–3487.
- Derosa, G.; Sahebkar, A.; Maffioli, P. The role of various peroxisome proliferator-activated receptors and their ligands in clinical practice. J. Cell. Physiol. 2018, 233, 153–161.
- Dong, X.; Tan, P.; Cai, Z.; Xu, H.; Li, J.; Ren, W.; Xu, H.; Zuo, R.; Zhou, J.; Mai, K.; et al. Regulation of FADS2 transcription by SREBP-1 and PPAR-α influences LC-PUFA biosynthesis in fish. Sci. Rep. 2017, 7.
- Norris, G.H.; Porter, C.M.; Jiang, C.; Millar, C.L.; Blesso, C.N. Dietary sphingomyelin attenuates hepatic steatosis and adipose tissue inflammation in high-fat-diet-induced obese mice. J. Nutr. Biochem. 2017, 40, 36–43.
- Maurizi, G.; Della Guardia, L.; Maurizi, A.; Poloni, A. Adipocytes properties and crosstalk with immune system in obesity-related inflammation. J. Cell. Physiol. 2018, 233, 88–97.
- Shi, Y.; Sun, X.; Sun, Y.; Hou, L.; Yao, M.; Lian, K.; Li, J.; Lu, X.; Jiang, L. Elevation of cortical C26:0 due to the decline of peroxisomal β-oxidation potentiates amyloid β generation and spatial memory deficits via oxidative stress in diabetic rats. Neuroscience 2016, 315, 125–135.
- Van Veldhoven, P.P. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res. 2010, 51, 2863–2895.
- Yagita, Y.; Shinohara, K.; Abe, Y.; Nakagawa, K.; Al-Owain, M.; Alkuraya, F.S.; Fujiki, Y. Deficiency of a retinal dystrophy protein, acyl-CoA binding domain-containing 5 (ACBD5), impairs peroxisomal β-oxidation of very-long-chain fatty acids. J. Biol. Chem. 2017, 292, 691–705.
- Kanamori, S.; Ishida, H.; Yamamoto, K.; Itoh, T. Construction of a series of intermediates in the β-oxidation pathway from THA to EPA via DHA in free acid form. Bioorg. Med. Chem. 2018, 26, 4390–4401.
- Zheng, F.; Cai, Y. Concurrent exercise improves insulin resistance and nonalcoholic fatty liver disease by upregulating PPAR-γ and genes involved in the beta-oxidation of fatty acids in ApoE-KO mice fed a high-fat diet. Lipids Health Dis. 2019, 18, 6.
- Le Cras, T.D.; Mobberley-Schuman, P.S.; Broering, M.; Fei, L.; Trenor, C.C., 3rd; Adams, D.M. Angiopoietins as serum biomarkers for lymphatic anomalies. Angiogenesis 2017, 20, 163–173.
- Périchon, R.; Bourre, J.M. Peroxisomal b-oxidation activity and catalase activity during development and aging in mouse liver. Biochimie 1995, 77, 288–293.
- An, H.J.; Lee, B.; Kim, S.M.; Kim, D.H.; Chung, K.W.; Ha, S.G.; Park, K.C.; Park, Y.J.; Kim, S.J.; Yun, H.Y.; et al. A PPAR pan agonist, MHY2013 alleviates age-related hepatic lipid accumulation by promoting fatty acid oxidation and suppressing inflammation. Biol. Pharm. Bull. 2018, 41, 29–35.
- Sanguino, E.; Roglans, N.; Alegret, M.; Sánchez, R.M.; Vázquez-Carrera, M.; Laguna, J.C. Atorvastatin reverses age-related reduction in rat hepatic PPARalpha and HNF-4. Br. J. Pharm. 2005, 145, 853–861.
- Chee, C.; Shannon, C.E.; Burns, A.; Selby, A.L.; Wilkinson, D.; Smith, K.; Greenhaff, P.L.; Stephens, F.B. Relative contribution of intramyocellular lipid to whole body fat oxidation is reduced with age, but subsarcolemmal lipid accumulation and insulin resistance are only associated with overweight individuals. Diabetes 2016, 65, 840–850.
- O’Brien, P.D.; Hinder, L.M.; Callaghan, B.C.; Feldman, E.L. Neurological consequences of obesity. Lancet Neurol. 2017, 16, 465–477.
- Okereke, O.I.; Rosner, B.A.; Kim, D.H.; Kang, J.H.; Cook, N.R.; Manson, J.E.; Buring, J.E.; Willett, W.C.; Grodstein, F. Dietary fat types and 4-year cognitive change in community-dwelling older women. Ann. Neurol. 2012, 72, 124–134.
- Pessayre, D.; Berson, A.; Fromenty, B.; Mansouri, A. Mitochondria in steatohepatitis. Semin. Liver Dis. 2001, 21, 57–69.
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472.
- Pessayre, D.; Mansouri, A.; Fromenty, B. Nonalcoholic steatosis and steatohepatitis. Mitochondrial dysfunction in steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G193–G199.
- Matsuzawa-Nagata, N.; Takamura, T.; Ando, H.; Nakamura, S.; Kurita, S.; Misu, H.; Ota, T.; Yokoyama, M.; Honda, M.; Miyamoto, K.; et al. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism 2008, 57, 1071–1077.
- Dalvi, P.S.; Chalmers, J.A.; Luo, V.; Han, D.-Y.D.; Wellhauser, L.; Liu, Y.; Tran, D.Q.; Castel, J.; Luquet, S.; Wheeler, M.B.; et al. High fat induces acute and chronic inflammation in the hypothalamus: Effect of high-fat diet, palmitate and TNF-α on appetite-regulating NPY neurons. Int. J. Obes. 2017, 41, 149–158.
- Tapias, V.; Hu, X.; Luk, K.C.; Sanders, L.H.; Lee, V.M.; Timothy Greenamyre, J. Synthetic alpha-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cell. Mol. Life Sci. 2017, 74, 2851–2874.
- Pistell, P.J.; Morrison, C.D.; Gupta, S.; Knight, A.G.; Keller, J.N.; Ingram, D.K.; Bruce-Keller, A.J. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J. Neuroimmunol. 2010, 219, 25–32.
- Ferreira, M.R.; Alvarez, S.M.; Illesca, P.; Giménez, M.S.; Lombardo, Y.B. Dietary Salba (Salvia hispanica L.) ameliorates the adipose tissue dysfunction of dyslipemic insulin-resistant rats through mechanisms involving oxidative stress, inflammatory cytokines and peroxisome proliferator-activated receptor γ. Eur. J. Nutr. 2018, 57, 83–94.
- Ana, C.; Danila, D.R.; Ana, M.; Carmen, G.; Lidia, M.; Mariano, R.-G.; Nuria, D.O. Inhibition of hippocampal long-term potentiation by high-fat diets: Is it related to an effect of palmitic acid involving glycogen synthase kinase-3? Neuroreport 2017, 28, 354–359.
- McLean, F.H.; Grant, C.; Morris, A.C.; Horgan, G.W.; Polanski, A.J.; Allan, K.; Campbell, F.M.; Langston, R.F.; Williams, L.M. Rapid and reversible impairment of episodic memory by a high-fat diet in mice. Sci. Rep. 2018, 8, 11976.
- Duffy, C.M.; Hofmeister, J.J.; Nixon, J.P.; Butterick, T.A. High fat diet increases cognitive decline and neuroinflammation in a model of orexin loss. Neurobiol. Learn. Mem. 2019, 157, 41–47.
- Woodie, L.; Blythe, S. The differential effects of high-fat and high-fructose diets on physiology and behavior in male rats. Nutr. Neurosci. 2018, 21, 328–336.
- Stranahan, A.M.; Norman, E.D.; Lee, K.; Cutler, R.G.; Telljohann, R.S.; Egan, J.M.; Mattson, M.P. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus 2008, 18, 1085–1088.
- Greenwood, C.E.; Winocur, G. High-fat diets, insulin resistance and declining cognitive function. Neurobiol. Aging 2005, 26, 42–45.
- Jena, P.K.; Sheng, L.; Di Lucente, J.; Jin, L.-W.; Maezawa, I.; Yvonne Wan, Y.-J. Dysregulated bile acid synthesis and dysbiosis are implicated in Western diet-induced systemic inflammation, microglial activation, and reduced neuroplasticity. FASEB J. 2018, 32, 2866–2877.
- Holloway, C.J.; Cochlin, L.E.; Emmanuel, Y.; Murray, A.; Codreanu, I.; Edwards, L.M.; Szmigielski, C.; Tyler, D.J.; Knight, N.S.; Saxby, B.K.; et al. A high-fat diet impairs cardiac high-energy phosphate metabolism and cognitive function in healthy human subjects. Am. J. Clin. Nutr. 2011, 93, 748–755.
- Edwards, L.M.; Murray, A.J.; Holloway, C.J.; Carter, E.E.; Kemp, G.J.; Codreanu, I.; Brooker, H.; Tyler, D.J.; Robbins, P.A.; Clarke, K. Short-term consumption of a high-fat diet impairs whole-body efficiency and cognitive function in sedentary men. FASEB J. 2011, 25, 1088–1096.
- Mittal, K.; Katare, D.P. Shared links between type 2 diabetes mellitus and Alzheimer’s disease: A review. Diabetes Metab. Syndr. Clin. Res. Rev. 2016, 10, S144–S149.
- Bernard, N.D.; Bunner, A.E.; Agarwal, U. Saturated and trans fats and dementia: A systematic review. Neurobiol. Aging 2014, 35, S65–S73.
- Solfrizzi, V.; Colacicco, A.M.; D’Introno, A.; Capurso, C.; Del Parigi, A.; Capurso, S.A.; Argentieri, G.; Capurso, A.; Panza, F. Dietary fatty acids intakes and rate of mild cognitive impairment. The Italian Longitudinal Study on Aging. Exp. Gerontol. 2006, 41, 619–627.
- Cherbuin, N.; Anstey, K.J. The Mediterranean diet is not related to cognitive change in a large prospective investigation: The PATH through life study. Am. J. Geriatr. Psychiatry 2012, 20, 635–639.
- Roberts, R.O.; Roberts, L.A.; Geda, Y.E.; Cha, R.H.; Pankratz, V.S.; O’Connor, H.M.; Knopman, D.S.; Petersen, R.C. Relative intake of macronutrients impacts risk of mild cognitive impairment or dementia. J. Alzheimer’s Dis. 2012, 32, 329–339.
- Engelhart, M.J.; Geerlings, M.I.; Ruitenberg, A.; Van Swieten, J.C.; Hofman, A.; Witteman, J.C.; Breteler, M.M. Diet and risk of dementia: Does fat matter? the Rotterdam Study. Neurology 2002, 59, 1915–1921.
- Luchsinger, J.A.; Tang, M.X.; Shea, S.; Mayeux, R. Caloric intake and the risk of Alzheimer disease. Arch. Neurol. 2002, 59, 1258–1263.
- Swanson, D.; Block, R.; Mousa, S.A. Omega-3 fatty acids EPA and DHA: Health benefits throughout life. Adv. Nutr. 2012, 3, 1–7.
- Luchtman, D.W.; Song, C. Cognitive enhancement by omega-3 fatty acids from childhood to old age: Findings from animal and clinical studies. Neuropharmacol. 2013, 64, 550–565.
- Devore, E.E.; Grodstein, F.; van Rooij, F.J.; Hofman, A.; Rosner, B.; Stampfer, M.J.; Witteman, J.C.; Breteler, M.M. Dietary intake of fish and omega-3 fatty acids in relation to long-term dementia risk. Am. J. Clin. Nutr. 2009, 90, 170–176.
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izqur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Invest. 2012, 122, 153–162.
- Wang, X.; Ge, A.; Cheng, M.; Guo, F.; Zhao, M.; Zhou, X.; Liu, L.; Yang, N. Increased hypothalamic inflammation associated with the susceptibility to obesity in rats exposed to high-fat diet. Exp. Diabetes Res. 2012, 2012.
- Andre, C.; Dinel, A.L.; Ferreira, G.; Laye, S.; Castanon, N. Diet-induced obesity progressively alters cognition, anxiety-like behavior and lipopolysaccharide-induced depressive-like behaviour: Focus on brain indoleamine 2,3-dioxygenase activation. Brain Behav. Immunol. 2014, 41, 10–21.
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharm. 2015, 74, 101–110.
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231.
- Wang, C.H.; Wu, S.B.; Wu, Y.T.; Wei, Y.H. Oxidative stress response elicited by mitochondrial dysfunction: Implication in the pathophysiology of aging. Exp. Biol. Med. 2013, 238, 450–460.
- Guo, C.Y.; Sun, L.; Chen, X.P.; Zhang, D.S. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014.
More
Information
Subjects:
Nutrition & Dietetics
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
13 Aug 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No