 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Steliana Ghibu | + 6453 word(s) | 6453 | 2021-08-05 04:16:21 | | | |
| 2 | Bruce Ren | -21 word(s) | 6432 | 2021-08-12 11:03:08 | | |
Video Upload Options
Cardiovascular complications are rapidly emerging as a major peril in COVID-19 in addition to respiratory disease. The mechanisms underlying the excessive effect of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection on patients with cardiovascular comorbidities remain only partly understood. SARS-CoV-2 infection is caused by binding of the viral surface spike (S) protein to the human angiotensin-converting enzyme 2 (ACE2), followed by the activation of the S protein by transmembrane protease serine 2 (TMPRSS2). ACE2 is expressed in the lung (mainly in type II alveolar cells), heart, blood vessels, small intestine, etc., and appears to be the predominant portal to the cellular entry of the virus. Based on current information, most people infected with SARS-CoV-2 virus have a good prognosis, while a few patients reach critical condition, especially the elderly and those with chronic underlying diseases. The “cytokine storm” observed in patients with severe COVID-19 contributes to the destruction of the endothelium, leading to “acute respiratory distress syndrome” (ARDS), multiorgan failure, and death. At the origin of the general proinflammatory state may be the SARS-CoV-2-mediated redox status in endothelial cells via the upregulation of ACE/Ang II/AT1 receptors pathway or the increased mitochondrial reactive oxygen species (mtROS) production. Furthermore, this vicious circle between oxidative stress (OS) and inflammation induces endothelial dysfunction, endothelial senescence, high risk of thrombosis and coagulopathy. The microvascular dysfunction and the formation of microthrombi in a way differentiate the SARS-CoV-2 infection from the other respiratory diseases and bring it closer to cardiovascular diseases like myocardial infarction and stroke. Due the role played by OS in the evolution of viral infection and in the development of COVID-19 complications, the use of antioxidants as adjuvant therapy seems appropriate in this new pathology. Alpha-lipoic acid (ALA) could be a promising candidate that, through its wide tissue distribution and versatile antioxidant properties, interferes with several signaling pathways.
1. Introduction
2. ACE2-Mediated SARS-CoV-2 Viral Toxicity
3. Endothelial Cell Damage: Endothelialitis
4. COVID-19 and “Cytokine Storm”
In addition to all disturbances already mentioned, the “cytokine storm” observed in patients with severe COVID-19 contributes to further destruction of the endothelium, leading to ARDS and multiorgan failure. Inflammation and “cytokine storm syndrome”, resembling hemophagocytic lymphohistiocytosis (HLH)/macrophage activation syndrome (MAS), leads to extreme morbidity and mortality due to the uncontrolled activation and proliferation of lymphocytes and macrophages. MAS has been reported in deaths related to autoimmune disorders and infections that are primarily of viral origin [26][27].
The term “cytokine storm” was first coined in 1993 to describe graft versus host diseases, and it is characterized by an uncontrolled and unrestricted production of pro-inflammatory cytokines and by a systemic hyper-inflammatory reaction. The cytokines constitute an essential and important part of the body’s immune response, but a “cytokine storm” is nearly always pathogenic, because of its detrimental effects on the host. The term has since been extended to describe the similar sudden cytokine releases associated with autoimmune diseases, sepsis, cancers, acute immunotherapy responses, and infectious diseases [28].
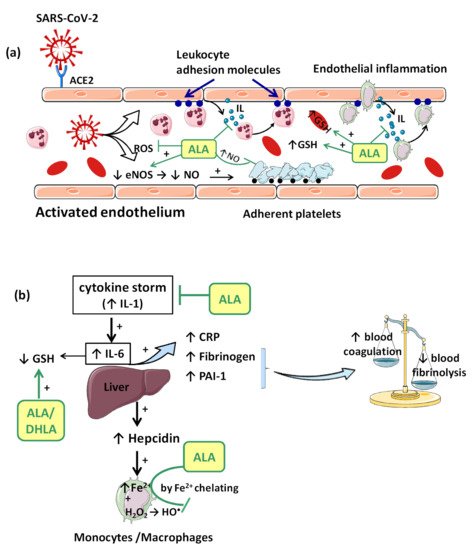
Consequences of viral infection of the respiratory epithelium include dysfunction and destruction of alveolar epithelium, and the increase in capillary endothelium permeability. These effects are mediated in part by the impact of the pro-inflammatory “cytokine storm” that is produced during the late antiviral innate immune response. The “cytokine storm” with a generalized hyperinflammatory state appears as a consequence of temporary failure of immune response mediated by type I interferons (IFNs) (e.g., IFN-α, IFN-β, etc.) during the initial period of SARS-CoV-2 infection [28]. In this context, IL-1 seems to represent the trigger factor to which IL-6 later joins [28]. IL-1 can induce its own gene expression, but it also stimulates the synthesis of other pro-inflammatory cytokines (TNF-α, IL-6) and chemokines, thus amplifying the cytokine overproduction and tissue infiltration of leukocytes. Moreover, IL-6 promotes an acute phase response characterized by increased hepatic synthesis of fibrinogen, plasminogen activator inhibitor-1 (PAI-1) and C reactive protein (CRP) (Figure 2b). Thus, besides affecting the integrity and function of endothelium, the “cytokine storm” is associated with a prothrombotic and antifibrinolytic imbalance with a hypercoagulable state responsible for venous thrombosis (blood clot) and for pulmonary embolism risk (Figure 2b) [24][28].
It has been reported that markers of systemic inflammation such as CRP, monocyte chemoattractant protein1 (MCP-1) and IL-6 are elevated in patients with poor clinical outcomes and in those with the need for mechanical ventilation, being correlated with the severity of pneumonia and the mortality rate [22][28].
Complementarily, pro-inflammatory cytokines such as TNF-α and IFN-γ are highly upregulated in COVID-19 patients, providing another amplification loop of “cytokine storm” and participating in cell death, tissue and organ damages [29][30].
5. COVID-19, Inflammation and Oxidative Stress
In COVID-19 patients, the imbalance of oxygen supply and demand caused by the inflammatory response and by endothelial dysfunction is similar to the pathophysiology of type 2 myocardial infarction [31]. However, the production of ROS and redox injuries is increased, exacerbating even further the pre-existing disorders, and potentially inducing chronic endothelial dysfunction [32].
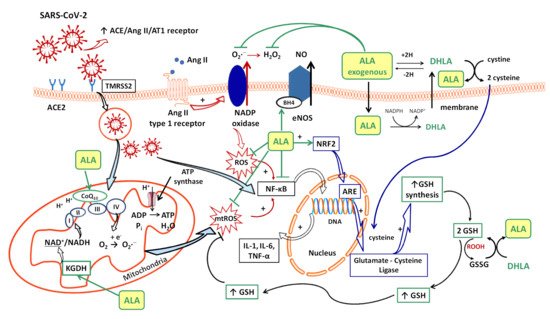
Several sources responsible for ROS synthesis have been reported in COVID-19; the starting point appears to be the endothelial cells, both by NADPH oxidase (NOx) and the electron leakage from the mitochondrial respiratory chain [4]. The anion superoxide (O2•−) produced via NOx, due the upregulation of Ang II/AT1 receptor pathway [33] together with the hydrogen peroxide (H2O2) and peroxynitrite anion (ONOO−) subsequently formed, or the direct interactions between viral and mitochondrial proteins, initiates a mitochondrial electron transport chain dysregulation (complex I and III) with a significant increase in mitochondrial ROS production [4]. Furthermore, this excess of ROS triggers the proinflammatory cytokine synthesis (IL-1β, IL-6, IL-18, TNF-α) through activation of NF-κB factor [4] and promotes a proinflammatory endothelial status (Figure 1).
In consequence, it is reasonable to assume that the endothelium contributes to COVID-19-associated vascular inflammation, particularly endothelialitis, in the various organs, endothelial cells being a key player in this new pathology. Moreover, through the endothelial cells, the inflammatory cascade promotes leukocyte recruitment and amplifies the local OS. Leukocyte transmigration is an important occurrence in the inflammatory response and involves the recruitment of circulating leukocytes, their adhesion to the endothelial cells, and diapedesis toward damaged tissues (Figure 2a). The immune response through increase in leukocyte activity fuels up the vascular ROS synthesis even if these highly reactive structures also play an important role as cell-signaling molecules for innate immunity and for maintaining endothelial homeostasis [4]. The enhanced ROS synthesis also impairs the local antioxidant defense, as highlighted by deacreases in superoxide dismutase (SOD), glutathione peroxidase (GPx), haem oxygenase activities, and reduced glutathione (GSH) levels [24], and exacerbates the general OS.
Accumulation of mononuclear cells (e.g., monocytes/macrophages system) in the small lung vessels is implicated in endothelial injury [25]. Endothelial cell dysfunction includes the impairment of local metabolic environment with a modification in the production of the vasodilator NO and ROS associated with an upregulation of leukocyte adhesion molecules (E-Selectin, P-Selectin) or intercellular adhesion molecule-1 (ICAM-1). Endothelial exocytosis initiates leukocyte and platelet adherence to the capillary wall and leads not only to vascular inflammation, but also to microthrombosis and microvascular obstruction (Figure 2a) [22].
Furthermore, platelet–neutrophil interaction and macrophage activation promote proinflammatory responses including “cytokine storm” and the formation of neutrophil extracellular traps (NETs). High levels of NETs have been reported in hospitalized patients with COVID-19. NETs induce endothelial injury and stimulate both extrinsic and intrinsic coagulation pathways with thrombin activation and clot formation, which amplifies microvascular dysfunctions [34].
Complex interactions between enzymatic activities, erythropoiesis, iron metabolism, hepcidin (a key regulator of the intracellular iron release), and growth/differentiation factor 15 (GDF15) have been demonstrated during the inflammatory process [35]. Within hours from bacterial and viral infections or other inflammatory stimuli, plasma iron concentrations decrease. This response is referred to as “hypoferremia of inflammation” and has been documented in humans. As we reported previously, the common mechanism of hypoferremia of inflammation is a cytokine-driven increase in hepcidin that downregulates ferroportin, and thereby decreases iron flow into extracellular fluid (Figure 2b). Interestingly, although most bacterial and viral infections rapidly increase hepcidin production in humans and mouse models [36], potential mechanisms of the systemic clinical findings of COVID-19 include the dysregulated iron homeostasis, resulting in OS and inflammatory response. Dysregulation of iron homeostasis with higher iron levels may support the progression of viral infections. Evaluating serum ferritin levels in COVID-19 patients may help to predict the outcome of this pathology. Trends and modifications of iron parameters are reported in many clinical studies, ferritin being a very early and non-specific indicator of inflammation [37].
Additionally, H2O2 formed by different pathways can diffuse across cell membranes, interacting with intracellular iron and inducing the synthesis of hydroxyl radical (HO•), one of the most reactive ROS, enhancing OS and oxidative injuries in COVID-19 patients [4]. The HO• formed can further induce telomeric DNA strand breaks with exacerbation of cellular senescence and the occurrence of all local perturbation derived from it (cellular inflammation, endothelial dysfunction, increased cellular susceptibility to the virus, etc.) [4].
6. Therapeutic Perspectives
Previous reviews have enumerated the potential therapeutic targets and strategies, both conventional and alternative, along with vaccine candidates in clinical trials against COVID-19. However, successful completion of drug development may require several years with no guarantee. Alternatively, already-established drugs can be repurposed to treat the COVID-19 infection. The anti-inflammatory treatment is as important as the antiviral treatment in COVID-19 management [4][28]. Therapies that target the immune response and decrease the cytokine storm in COVID-19 patients have become an application for new clinical trials [38]. Moreover, antithrombotic treatment, especially anticoagulant agents, is provided to prevent micro- or macro-thromboses and microvascular obstructions.
Due the fact that the OS establishes the favorable conditions for the virus’ entry into cells and intracellular virus replication, and is also closely related to the proinflammatory status and endothelial dysfunction, it could be considered an important therapeutic target in the fight against this new coronavirus. Generally, by modulating the endogenous redox status, the immune response could be regulated, too [33]. In this regard, priority should be given to antioxidants whose safety profile has already been proven in experimental models or clinical trials, and alpha-lipoic acid (ALA) could be an appropriate and promising candidate for this new therapeutic approach.
6.1. Alpha-Lipoic Acid, Antioxidant and Endothelial Protection Effects
ALA or thioctic acid (5-(1,2-dithiolan-3-yl) pentanoic acid) is a potent and complex antioxidant compound with a very good safety profile [39][40]. Moreover, in the last few years, ALA has exceeded the status of antioxidant, its ability to mediate and regulate multiple signaling pathways having been demonstrated. Thus, besides its use as adjuvant therapy for diabetic neuropathy in some European countries, multiple studies have reported the benefits of ALA treatment in metabolic disorders (hyperglycemia, tissue insulin resistance, dyslipidemia or obesity) [41][42][43], endothelial dysfunction [44][45][46][47] or in various inflammatory processes [45][48].
Another great advantage of ALA is represented by its amphiphilic properties, due to which ALA can be distributed both in hydrophilic (plasma, cell cytoplasm, etc.) and in lipophilic (cell membranes, etc.) environments. Actually, the amphiphilic character of ALA is unique among antioxidants [49].
Under physiological conditions, the dietary intake of ALA (R-isomer) joins de novo ALA (R-isomer) synthesis, thus ensuring the proper functioning of basic physiological processes (cofactor of mitochondrial pyruvate dehydrogenase and α-ketoglutarate dehydrogenase (KGDH)) [39]. However, in pathological conditions, especially in complex situations associated with oxidative injuries and a high level of OS, the need for ALA treatment is justified and may be recommended. Depending on the severity of the disease and the state of consciousness of the patient, oral or parenteral (intravenous) pharmaceutical forms of ALA can be used, and the doses can be adjusted, with a daily administration between 600 and 1200 mg ALA frequently being recommended. After oral administration, ALA is rapidly absorbed from the gastrointestinal tract, followed by both its transport into tissues as well as its renal excretion, which explains its very short half-life (30–40 minutes after oral administration and 12 minutes after intravenous administration) [50].
Intracellularly (endothelial cells, erythrocytes, etc.), ALA is reduced to dihydrolipoic acid (DHLA, 6,8-dimercapto-6λ3-octoanoic acid), which is subsequently extracellularly released and oxidized to ALA. DHLA presents superior antioxidant properties compared to ALA [51] due to the two free thiol groups within its structure, and also contributes to the recycling and prolongation of ALA effects over a longer period of time (Figure 1) [49][52][53].
ALA is a complex antioxidant molecule that can interfere with several signaling pathways of OS. In addition to its ability to directly scavenge the reactive oxygen species (HO•, HClO, 1O2), the ALA/DHLA redox couple may indirectly provide antioxidant protection through transition metal chelation, especially of divalent metals (iron and copper), and regeneration of the reduced forms of some endogenous antioxidants (vitamin E, vitamin C and glutathione) (Figure 1, Figure 2a,b) [54][49][39]. Unlike the ions in the extracellular fluids, the intracellular iron is more labile and would be more easily involved in redox reactions [55]. Additionally, metal ions such as iron and copper catalyze the electron transfer from one oxygen species to another [56]. Although Fe2+ is involved in various physiological processes, in the presence of H2O2, a mild oxidizing agent, it has great potential to generate, by Fenton reaction, more aggressive free radicals such as HO•, and thus enhances the OS. ALA cannot neutralize H2O2, but by chelating the low iron concentration, it can prevent the oxidative injury [57]. Both ALA and DHLA have metal-chelating capacity, ALA reacts with Fe2+ while DHLA chelates even Fe3+ [56][57]. Moreover, by chelating Cu2+, the ALA/DHLA system prevents copper-induced lipid peroxidation or Cu2+-catalyzed ascorbic acid oxidation with catalase inactivation and increased production of H2O2 within the erythrocytes [58].
Due to these properties, the ALA/DHLA system could attenuate the erythrocyte oxidative injuries and cell lysis derived from these, especially in a context of increased plasma OS, as might be found in COVID-19. Nevertheless, to obtain maximum effectiveness in all aspects of the COVID-19 treatment, the immunity stimulation adjuvant therapy that is frequently used, and which may include high doses of vitamin C and zinc, should be taken into account [59][60]. Thus, in order to avoid drug interactions such as gastrointestinal or plasmatic Zn2+ chelation by ALA, it is mandatory to space out the administration of the two pharmaceutical products, possibly with the initial administration of ALA, especially since ALA presents the advantage of a single daily administration and a short half-life.
Furthermore, due to its high redox potential (−320 mV), the ALA/DHLA system provides more protection from oxidative damage, being more effective than the endogenous reduced/oxidized glutathione (GSH/GSSG) system (−240 mV), a basic constituent of the first line of antioxidant defense [54][49].
The vascular endothelium seems to be a privileged target for the protective action of ALA. By reducing OS, ALA can restore the endothelial nitric oxide synthase (eNOS) activity with the consequent increase of NO bioavailability and improvement in endothelial function [61]. This is partially done by preventing the oxidative depletion of tetrahydrobiopterin (BH4), an essential cofactor of eNOS (Figure 1) [54][62], and partially by increasing the activity of dimethylarginine dimethylaminohydrolase (DDAH), the metabolizing enzyme of plasma asymmetric dimethylarginine (ADMA) [63]. In this way, on the one hand, eNOS is recoupled, and its function is switched to resuming NO production instead of superoxide anion production [62], while, on the other hand, eNOS inhibition by the endogenous ADMA is canceled [63]. By enhancing NO and decreasing ONOO− levels, the nitric-oxide-dependent vascular relaxation is preserved, and platelet aggregation is limited, too (Figure 2a) [54][64]. Thus, it has been reported that the flow-mediated dilation (FMD) level, an accurate, non-invasive index of endothelial function, was significantly improved under ALA treatment [45][47][61], with the greatest effect being obtained in diabetic patients [44][61].
6.2. Alpha-Lipoic Acid Effects in COVID-19
ALA is an antioxidant agent with great prospects for use as adjuvant therapy in COVID-19 patients. More precisely, besides reducing OS and protecting the vascular endothelium, it can diminish the cellular entry of SARS-CoV-2 and the inflammatory process, and it can indirectly stimulate the immune system.
Some studies have mentioned that the disulfide–thiol balance would play an important role in cell–virus interaction processes and the virus’ entry into the cells. OS significantly influences this ratio and the viral infection rate [33]. Thus, ROS can easily oxidize the cysteine residues from S protein (RBD) and membrane ACE2 (peptidase domain), leading to enhanced numbers of disulfide groups and the increase in affinity between the two structures, even with a possible increase in COVID-19 severity in certain situations [33][65]. As we reported previously, the relationship between the ALA/DHLA system and thiol redox status has already been established. On the one hand, it increases the GSH/GSSG ratio significantly, and, by modulation of the intracellular HS-/-S-S- ratio, it activates the insulin-signaling pathway with the increase of intracellular glucose uptake and reduces glycemia [49]. Thus, it is possible that through a similar mechanism, the cell entry of SARS-CoV-2 could be affected in the presence of ALA treatment.
Unfortunately, there are very few data on the effects of ALA as adjuvant therapy in patients with COVID-19; most publications related to ALA and different aspects of COVID-19 are reviews. However, in a small recent study, Zhong et al. evaluated the clinical efficacy and safety of ALA in patients with critical forms of COVID-19. Although the study was performed on a small number of patients and the intravenous treatment with ALA (once daily dose of 1200 mg) was administered only for 7 days, in addition to the standard medical care, the results obtained show a lower all-cause mortality rate in the ALA COVID-19 group than in the placebo group, at 30-day follow-up [66]. Additionally, in a previous study, it was demonstrated in vitro that the enhanced susceptibility to viral infection with Human Coronavirus (HCoV) 229E in glucose-6-phosphate dehydrogenase (G6PD)-deficient cells was ameliorated by ALA; this strengthens even further the hypothesis that a high cellular OS provides a favorable environment for viral replication and virulence, and highlights the role of antioxidants, especially of GSH, in innate immunity [67]. Further studies with a larger patient cohort are needed to highlight and validate the role of ALA in patients with different forms of COVID-19.
Another important effect of ALA, both dependent on its antioxidant properties, as well as independent of them, is the anti-inflammatory effect. By reducing the ROS, the general inflammatory process is also decreased. Moreover, by chelating copper, the ALA/DHLA system inhibits the activation and nuclear translocation of NF-κB, with the decrease in pro-inflammatory cytokine secretion, regardless of its antioxidant properties (Figure 1) [49][39][68]. Due to the low cytokine levels and by inducing heme oxygenase-1 upregulation, ALA subsequently decreases the adhesion molecules’ expression (E-selectine, vascular cell adhesion molecule-1 (VCAM-1), intracellular adhesion molecule-1 (ICAM-1)) and the extravascular leucocyte migration (Figure 2a) [69][70]. A significant number of recent clinical trials [45][48][71] and experimental studies [42][43] have reported the efficacy of ALA in different inflammatory process by decreasing the inflammatory markers such us CRP, IL-6 or TNF-α [72]. In this context, it was noted that ALA contributes to multiple organ protection in sepsis [72], in part through activation of autophagy, too [73].
A relevant contribution of mitochondrial ROS synthesis and mitochondrial dysfunction in COVID-19 pathogenesis has been noted. Thus, some studies have proposed mitochondria to be among the main adjuvant therapeutic targets in sepsis [74] and other acute disorders, including in COVID-19 management [72][75]. ALA is a cofactor of pyruvat dehydrogenase and α-ketoglutarate dehydrogenase (KGDH, the E2 sub-unit), key regulatory enzymes within the Krebs cycle, which ultimately influences the adenosine triphosphate (ATP) production [53][76]. KGDH is a key sensor of mitochondrial redox status, which in turn is inactivated by an elevated level of ROS; it is also the key rate-limiting enzyme for nicotinamide adenine dinucleotide (NADH) production, thus controlling the supply with reducing equivalents or mitochondrial electron transports (Figure 1) [76][77]. A decline in KGDH activity has been reported in a variety of neurological and cardiovascular disorders associated with OS. Moreover, a specific inhibition of KGDH through oxidative reactive products can induce mitochondrial cytochrome c release and cell death [76][77]. Thus, both through its complex antioxidant properties and as a cofactor of KGDH [49][53], ALA could mitigate mitochondrial oxidative damages and improve mitochondrial function with the maintaining of tissues’ homeostasis in all of the above-mentioned pathologies [53][72][74]. Moreover, the ALA/DHLA system is able to reduce ubiquinone (CoQ10) to ubiquinol, an important constituent of the mitochondrial electron transport chain, and to enhance the NAD(P)H/NAD(P)+ ratio, a component of cell redox state (Figure 1) [53][64].
Glutathione, which is implicated in redox state, is an essential endogenous antioxidant responsible for reducing hydroperoxides and H2O2 and for preventing cellular oxidative damage. A decrease in circulating or intracellular GSH or GSH/GSSG ratio has been reported in different bacterial or viral infections, in lung function impairment, and in cardiometabolic disorders [78]. Generally, GSH depletion is a direct consequence of viral infection; this aspect is required for virus replication [78]. More precisely, the loss of GSH affects Na+ H+ membrane antiport with the decrease in intracellular pH, which facilitates both virus endocytosis and its replication. Secondarily, the viruses possess a variety of adaptive mechanisms (activation of NADPH oxidase and NF-κB or down regulation of NRF2 expression), which further reduce the host cell GSH. The GSH deficiency in SARS-CoV2 infection is firstly due to a deficient nuclear translocation of NRF2 and an elevated level of IL-6. An inverse relationship between IL-6 and GHS levels has been reported in COVID-19 patients [78]. Subsequently, a low level of GSH can be responsible for immune dysfunctions (critical activity of natural killer lymphocytes), an increased of viral load and a high susceptibility to viral infection [33]. At the opposite pole, a high level of glutathione is associated with leukocyte proliferation, strengthened immune system, and an effective antiviral protection. By maintaining an endogen high GSH/GSSG ratio, virus replication and severe forms of COVID-19 can be prevented [33]. Thus, an antioxidant agent that keeps or enhances reduced glutathione levels could prevent lymphocyte exhaustion or immune cell depletion due to a chronic hyperinflammation state or to a cytokine excess [78]. Although encouraging results have been obtained with N-acetyl cysteine (NAC) or exogenous GSH, ALA could successfully complete the list of molecules with an important effect on GSH in COVID-19 infection. However, it should taken into account that NAC presents a low cerebral bioavailability, and intracellular delivery of exogenous GSH may be deficient in some tissues (e.g., heart, brain, etc.) due to the lack of specific carriers [64]. In addition to a wide tissue distribution, ALA is a versatile antioxidant that can enhance GSH/GSSG ratio by several mechanisms. The ALA/DHLA system stimulates GSH synthesis by increasing cellular cysteine uptake, a rate-limiting substrate for GSH synthesis, and by activation of the NRF2-ARE signaling pathways with the increased activity of glutamate–cysteine ligase (the rate-limiting enzyme in GSH synthesis). Additionally, its ability to restore GSH from oxidized glutathione should not be neglected (Figure 1, Figure 2a,b) [49][53][64]. GSH levels decrease in relation to age and, additionally, age together with some comorbidities such as diabetes, obesity and hypertension have a major impact on the development of severe forms of COVID-19 [33][78].
Another important aspect of the ALA-GSH relationship that could be also capitalized upon in the context of SARS-CoV-2 infection is the ALA hepatoprotection in acetaminophen-induced hepatotoxicity. Acetaminophen (paracetamol) is one of the most widely used and well-tolerated analgesic antipyretic drugs, but at overdoses, it can induce hepatic necrosis via its highly reactive intermediate metabolite, N-acetyl-para-benzoquinone imine (NAPQI). More specifically, in acetaminophen overdose or when glutathione is depleted by 70%, the excessive amount of NAPQI covalently binds to the hepatic intracellular protein sulfhydryl groups, including those from cytoplasmic or mitochondrial enzymes, and induces hepatic OS, cellular toxicity, and hepatocytolysis, which lead to irreversible hepatic necrosis [79][80]. In these situations, the administration of NAC is authorized, but there are also some experimental studies that highlight the hepatic benefits of ALA both by reducing local OS and by restoring the optimal level of GSH [81][82]. Thus, in patients with COVID-19 and a high dose acetaminophen-treated fever, which do not tolerate NAC or who are not recommended to take mucolytics (expectorants), ALA could be an alternative of NAC. Additionally, due to multiple ways to increase the GSH levels, but also due to multiple processes leading to GSH depletion, the hepatoprotective effect of ALA might be obvious or relevant after a period of successive administrations. Furthermore, administration of ALA during the same period of time as acetaminophen may provide maximum hepatic protection.
7. Conclusions
Besides respiratory symptoms, COVID-19 is also considered an endothelial and coagulopathy disorder with severe cardiovascular complications. Furthermore, the main connection between them seems to be the inflammation via mitochondrial and endothelial ROS production. In this context, the structures involved in ROS synthesis and the multiple signaling pathways of OS may be a justified therapeutic target in COVID-19 management. Through the various possibilities of modulating the cellular redox state, ALA could be a potential adjuvant therapy in COVID-19 patients. More randomized control trials are needed to evaluate and confirm the efficacy of ALA in COVID-19 infection.
References
- Huppert, L.A.; Matthay, M.A.; Ware, L.B. Pathogenesis of Acute Respiratory Distress Syndrome. Semin. Respir. Crit. Care Med. 2019, 40, 31–39.
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558.
- Han, H.; Yang, L.; Liu, R.; Liu, F.; Liu, F.; Wu, K.L.; Li, J.; Liu, X.H.; Zhu, C.L. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin. Chem. Lab. Med. 2020, 58, 1116–1120.
- Chang, R.; Mamun, A.; Dominic, A.; Le, N.T. SARS-CoV-2 Mediated Endothelial Dysfunction: The Potential Role of Chronic Oxidative Stress. Front. Physiol. 2021, 11.
- Kotta, S.; Aldawsari, H.M.; Badr-Eldin, S.M.; Alhakamy, N.A.; Md, S.; Nair, A.B.; Deb, P.K. Combating the Pandemic COVID-19: Clinical Trials, Therapies and Perspectives. Front. Mol. Biosci. 2020, 7.
- Malik, S.; Gupta, A.; Zhong, X.; Rasmussen, T.P.; Manautou, J.E.; Bahal, R. Emerging therapeutic modalities against covid-19. Pharmaceuticals 2020, 13, 188.
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signalin g in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023.
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S. wen Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149.
- Trougakos, I.P.; Stamatelopoulos, K.; Terpos, E.; Tsitsilonis, O.E.; Aivalioti, E.; Paraskevis, D.; Kastritis, E.; Pavlakis, G.N.; Dimopoulos, M.A. Insights to SARS-CoV-2 life cycle, pathophysiology, and rationalized treatments that target COVID-19 clinical complications. J. Biomed. Sci. 2021, 28, 9.
- Rochette, L.; Lorin, J.; Zeller, M.; Guilland, J.C.; Lorgis, L.; Cottin, Y.; Vergely, C. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: Possible therapeutic targets? Pharmacol. Ther. 2013, 140, 239–257.
- Clerkin, K.J.; Fried, J.A.; Raikhelkar, J.; Sayer, G.; Griffin, J.M.; Masoumi, A.; Jain, S.S.; Burkhoff, D.; Kumaraiah, D.; Rabbani, L.R.; et al. COVID-19 and Cardiovascular Disease. Circulation 2020, 141, 1648–1655.
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme: Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243.
- Karamyan, V.T.; Speth, R.C. Enzymatic pathways of the brain renin-angiotensin system: Unsolved problems and continuing challenges. Regul. Pept. 2007, 143, 15–27.
- Zhong, J.; Basu, R.; Guo, D.; Chow, F.L.; Byrns, S.; Schuster, M.; Loibner, H.; Wang, X.H.; Penninger, J.M.; Kassiri, Z.; et al. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation 2010, 122, 717–728.
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santo, M.J. The ACE2/Angiotensin-(1-7)/Mas axis of the renin-angiotensin system: Focus on Angiotensin-(1-7). Physiol. Rev. 2018, 98, 505–553.
- Oudot, A.; Vergely, C.; Ecarnot-Laubriet, A.; Rochette, L. Pharmacological concentration of angiotensin-(1-7) activates NADPH oxidase after ischemia-reperfusion in rat heart through AT1 receptor stimulation. Regul. Pept. 2005, 127, 101–110.
- Turner, A.J.; Hiscox, J.A.; Hooper, N.M. ACE2: From vasopeptidase to SARS virus receptor. Trends Pharmacol. Sci. 2004, 25, 291–294.
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687.
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Fasouli, E.S.; Samari, S.; et al. Cells and gene expression programs in the adult human heart. bioRxiv 2020.
- Song, H.; Seddighzadeh, B.; Cooperberg, M.R.; Huang, F.W. Expression of ACE2, the SARS-CoV-2 Receptor, and TMPRSS2 in Prostate Epithelial Cells. Eur. Urol. 2020, 78, 296–298.
- Vrints, C.J.M.; Krychtiuk, K.A.; Van Craenenbroeck, E.M.; Segers, V.F.; Price, S.; Heidbuchel, H. Endothelialitis plays a central role in the pathophysiology of severe COVID-19 and its cardiovascular complications. Acta Cardiol. 2020, 76, 109–124.
- Lowenstein, C.J.; Solomon, S.D. Severe COVID-19 Is a microvascular disease. Circulation 2020, 142, 1609–1611.
- Gladka, M.M.; Maack, C. The endothelium as Achilles’ heel in COVID-19 patients. Cardiovasc. Res. 2020, 116, e195–e197.
- Libby, P.; Lüscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044.
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418.
- Henderson, L.A.; Canna, S.W.; Schulert, G.S.; Volpi, S.; Lee, P.Y.; Kernan, K.F.; Caricchio, R.; Mahmud, S.; Hazen, M.M.; Halyabar, O.; et al. On the Alert for Cytokine Storm: Immunopathology in COVID-19. Arthritis Rheumatol. 2020, 72, 1059–1063.
- Teuwen, L.A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391.
- Kim, J.S.; Lee, J.Y.; Yang, J.W.; Lee, K.H.; Effenberger, M.; Szpirt, W.; Kronbichler, A.; Shin, J.I. Immunopathogenesis and treatment of cytokine storm in COVID-19. Theranostics 2020, 11, 316–329.
- Wang, H.; Luo, S.; Shen, Y.; Li, M.; Zhang, Z.; Dong, Y.; Zhou, H.; Lin, L.; Guo, W.; Kang, Z.; et al. Multiple Enzyme Release, Inflammation Storm and Hypercoagulability Are Prominent Indicators For Disease Progression A Multi-Centered Correlation Study with CT Imaging Score. SSRN Electron. J. 2020. Available online: https://papers.ssrn.com/sol3/papers.cfm?abstract_id=3544837 (accessed on 9 July 2021).
- Kam, Y.W.; Ahmed, M.Y.; Amrun, S.N.; Lee, B.; Refaie, T.; Elgizouli, K.; Fong, S.W.; Renia, L.; Ng, L.F.P. Systematic analysis of disease-specific immunological signatures in patients with febrile illness from Saudi Arabia. Clin. Transl. Immunol. 2020, 9, e1163.
- Musher, D.M.; Abers, M.S.; Corrales-Medina, V.F. Acute Infection and Myocardial Infarction. N. Engl. J. Med. 2019, 380, 171–176.
- Akki, R.; Fath, N.; Mohti, H. COVID-19: Oxidative Preconditioning as a Potential Therapeutic Approach. ACS Chem. Neurosci. 2020, 11, 3732–3740.
- Suhail, S.; Zajac, J.; Fossum, C.; Lowater, H.; McCracken, C.; Severson, N.; Laatsch, B.; Narkiewicz-Jodko, A.; Johnson, B.; Liebau, J.; et al. Role of Oxidative Stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) Infection: A Review. Protein J. 2020, 39, 644–656.
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179.
- Gudjoncik, A.; Guenancia, C.; Zeller, M.; Cottin, Y.; Vergely, C.; Rochette, L. Iron, oxidative stress, and redox signaling in the cardiovascular system. Mol. Nutr. Food Res. 2014, 58, 1721–1738.
- Michels, K.; Nemeth, E.; Ganz, T.; Mehrad, B. Hepcidin and Host Defense against Infectious Diseases. PLoS Pathog. 2015, 11.
- Notz, Q.; Schmalzing, M.; Wedekink, F.; Schlesinger, T.; Gernert, M.; Herrmann, J.; Sorger, L.; Weismann, D.; Schmid, B.; Sitter, M.; et al. Pro- and Anti-Inflammatory Responses in Severe COVID-19-Induced Acute Respiratory Distress Syndrome—An Observational Pilot Study. Front. Immunol. 2020, 11, 581338.
- Wang, J.; Jiang, M.; Chen, X.; Montaner, L.J. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J. Leukoc. Biol. 2020, 108, 17–41.
- Shay, K.P.; Moreau, R.F.; Smith, E.J.; Smith, A.R.; Hagen, T.M. Alpha-lipoic acid as a dietary supplement: Molecular mechanisms and therapeutic potential. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1149–1160.
- Derosa, G.; D’angelo, A.; Preti, P.; Maffioli, P. Safety and Efficacy of Alpha Lipoic Acid during 4 Years of Observation: A Retrospective, Clinical Trial in Healthy Subjects in Primary Prevention. Drug Des. Devel. Ther. 2020, 14, 5367–5374.
- Rahimlou, M.; Asadi, M.; Banaei Jahromi, N.; Mansoori, A. Alpha-lipoic acid (ALA) supplementation effect on glycemic and inflammatory biomarkers: A Systematic Review and meta- analysis. Clin. Nutr. ESPEN 2019, 32, 16–28.
- Pop, C.; Ștefan, M.G.; Muntean, D.M.; Stoicescu, L.; Gal, A.F.; Kiss, B.; Morgovan, C.; Loghin, F.; Rochette, L.; Lauzier, B.; et al. Protective effects of a discontinuous treatment with alpha-lipoic acid in obesity-related heart failure with preserved ejection fraction, in rats. Antioxidants 2020, 9, 1073.
- Ghibu, S.; Craciun, C.E.; Rusu, R.; Morgovan, C.; Mogosan, C.; Rochette, L.; Gal, A.F.; Dronca, M. Impact of Alpha-Lipoic Acid Chronic Discontinuous Treatment in Cardiometabolic Disorders and Oxidative Stress Induced by Fructose Intake in Rats. Antioxidants 2019, 8, 636.
- Lee, S.R.; Jeong, M.H.; Lim, S.Y.; Hong, S.N.; Kim, K.H.; Sohn, I.S.; Hong, Y.J.; Park, H.W.; Kim, J.H.; Kim, W.; et al. The effect of alpha lipoic acid (Thioctacid HR®) on endothelial function in diabetic and hypertensive patients. Korean Circ. J. 2006, 36, 559–564.
- Lai, S.; Petramala, L.; Muscaritoli, M.; Cianci, R.; Mazzaferro, S.; Mitterhofer, A.P.; Pasquali, M.; D’Ambrosio, V.; Carta, M.; Ansuini, M.; et al. α-lipoic acid in patients with autosomal dominant polycystic kidney disease. Nutrition 2020, 71, 110594.
- Zou, H.; Wang, H.; Liu, T.; Li, X.; Zhu, X.; Wang, Z. Protective role of α-lipoic acid in hyperuricemia-induced endothelial dysfunction. Exp. Ther. Med. 2017, 13, 3047–3054.
- Heinisch, B.B.; Francesconi, M.; Mittermayer, F.; Schaller, G.; Gouya, G.; Wolzt, M.; Pleiner, J. Alpha-lipoic acid improves vascular endothelial function in patients with type 2 diabetes: A placebo-controlled randomized trial. Eur. J. Clin. Investig. 2010, 40, 148–154.
- Saboori, S.; Falahi, E.; Eslampour, E.; Zeinali Khosroshahi, M.; Yousefi Rad, E. Effects of alpha-lipoic acid supplementation on C-reactive protein level: A systematic review and meta-analysis of randomized controlled clinical trials. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 779–786.
- Rochette, L.; Ghibu, S.; Muresan, A.; Vergely, C. Alpha-lipoic acid: Molecular mechanisms and therapeutic potential in diabetes. Can. J. Physiol. Pharmacol. 2015, 93, 1021–1027.
- Uchida, R.; Okamoto, H.; Ikuta, N.; Terao, K.; Hirota, T. Enantioselective pharmacokinetics of α-lipoic acid in rats. Int. J. Mol. Sci. 2015, 16, 22781–22794.
- Ghibu, S.; Lauzier, B.; Delemasure, S.; Amoureux, S.; Sicard, P.; Vergely, C.; Muresan, A.; Mogosan, C.; Rochette, L. Antioxidant properties of alpha-lipoic acid: Effects on red blood membrane permeability and adaptation of isolated rat heart to reversible ischemia. Mol. Cell. Biochem. 2009, 320, 141–148.
- Jones, W.; Li, X.; Qu, Z.C.; Perriott, L.; Whitesell, R.R.; May, J.M. Uptake, recycling, and antioxidant actions of α-lipoic acid in endothelial cells. Free Radic. Biol. Med. 2002, 33, 83–93.
- Packer, L.; Cadenas, E. Lipoic acid: Energy metabolism and redox regulation of transcription and cell signaling. J. Clin. Biochem. Nutr. 2011, 48, 26–32.
- Rochette, L.; Ghibu, S.; Richard, C.; Zeller, M.; Cottin, Y.; Vergely, C. Direct and indirect antioxidant properties of α-lipoic acid and therapeutic potential. Mol. Nutr. Food Res. 2013, 57, 114–125.
- Ghibu, S.; Richard, C.; Vergely, C.; Zeller, M.; Cottin, Y.; Rochette, L. Antioxidant properties of an endogenous thiol: Alpha-lipoic acid, useful in the prevention of cardiovascular diseases. J. Cardiovasc. Pharmacol. 2009, 54, 391–398.
- Biewenga, G.P.; Haenen, G.R.M.M.; Bast, A. The pharmacology of the antioxidant: Lipoic acid. Gen. Pharmacol. 1997, 29, 315–331.
- Muhammad, M.T.; Khan, M.N. Kinetics, mechanistic and synergistic studies of Alpha lipoic acid with hydrogen peroxide. J. Saudi Chem. Soc. 2017, 21, 123–131.
- Ou, P.; Tritschler, H.J.; Wolff, S.P. Thioctic (lipoic) acid: A therapeutic metal-chelating antioxidant? Biochem. Pharmacol. 1995, 50, 123–126.
- Kontoghiorghes, G.J.; Kolnagou, A.; Kontoghiorghe, C.N.; Mourouzidis, L.; Timoshnikov, V.A.; Polyakov, N.E. Trying to Solve the Puzzle of the Interaction of Ascorbic Acid and Iron: Redox, Chelation and Therapeutic Implications. Medicines 2020, 7, 45.
- Di Renzo, L.; Gualtieri, P.; Pivari, F.; Soldati, L.; Attinà, A.; Leggeri, C.; Cinelli, G.; Tarsitano, M.G.; Caparello, G.; Carrano, E.; et al. COVID-19: Is there a role for immunonutrition in obese patient? J. Transl. Med. 2020, 18, 415.
- Jalilpiran, Y.; Hajishafiee, M.; Khorshidi, M.; Rezvani, H.; Mohammadi-Sartang, M.; Rahmani, J.; Mousavi, S.M. The effect of Alpha-lipoic acid supplementation on endothelial function: A systematic review and meta-analysis. Phyther. Res. 2020, 35, 2386–2395.
- Schulz, E.; Wenzel, P.; Münzel, T.; Daiber, A. Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxidants Redox Signal. 2014, 20, 308–324.
- Lee, W.J.; Kim, S.H.; Kim, G.H.; Han, S.M.; Won, J.C.; Jung, C.H.; Park, H.S.; Choi, D.S.; Lee, K.U.; Park, J.Y. α-Lipoic acid activates dimethylarginine dimethylaminohydrolase in cultured endothelial cells. Biochem. Biophys. Res. Commun. 2010, 398, 653–658.
- Golbidi, S.; Badran, M.; Laher, I. Diabetes and alpha lipoic acid. Front. Pharmacol. 2011, 2, 69.
- Hati, S.; Bhattacharyya, S. Impact of Thiol−Disulfide Balance on the Binding of Covid-19 Spike Protein with Angiotensin-Converting Enzyme 2 Receptor. ACS Omega 2020, 5, 16292–16298.
- Zhong, M.; Sun, A.; Xiao, T.; Yao, G.; Sang, L.; Zheng, X.; Zhang, J.; Jin, X.; Xu, L.; Yang, W.; et al. A Randomized, single-blind, group sequential, active-controlled study to evaluate the clinical efficacy and safety of α-Lipoic acid for critically ill patients with coronavirus disease 2019 (COVID-19). medRxiv 2020.
- Wu, Y.-H.; Tseng, C.-P.; Cheng, M.-L.; Ho, H.-Y.; Shih, S.-R.; Chiu, D.T.-Y. Glucose-6-Phosphate Dehydrogenase Deficiency Enhances Human Coronavirus 229E Infection. J. Infect. Dis. 2008, 197, 812–816.
- ZHANG, W.-J.; FREI, B. α-Lipoic acid inhibits TNF-a-induced NF-κB activation and adhesion molecule expression in human aortic endothelial cells. FASEB J. 2001, 15, 2423–2432.
- Khalili, M.; Azimi, A.; Izadi, V.; Eghtesadi, S.; Mirshafiey, A.; Sahraian, M.A.; Motevalian, A.; Norouzi, A.; Sanoobar, M.; Eskandari, G.; et al. Does lipoic acid consumption affect the cytokine profile in multiple sclerosis patients: A double-blind, placebo-controlled, randomized clinical trial. Neuroimmunomodulation 2014, 21, 291–296.
- Chaudhary, P.; Marracci, G.H.; Bourdette, D.N. Lipoic acid inhibits expression of ICAM-1 and VCAM-1 by CNS endothelial cells and T cell migration into the spinal cord in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2006, 175, 87–96.
- Akbari, M.; Ostadmohammadi, V.; Tabrizi, R.; Mobini, M.; Lankarani, K.B.; Moosazadeh, M.; Heydari, S.T.; Chamani, M.; Kolahdooz, F.; Asemi, Z. The effects of alpha-lipoic acid supplementation on inflammatory markers among patients with metabolic syndrome and related disorders: A systematic review and meta-analysis of randomized controlled trials. Nutr. Metab. 2018, 15, 39.
- Pagano, G.; Manfredi, C.; Pallardó, F.V.; Lyakhovich, A.; Tiano, L.; Trifuoggi, M. Potential roles of mitochondrial cofactors in the adjuvant mitigation of proinflammatory acute infections, as in the case of sepsis and COVID-19 pneumonia. Inflamm. Res. 2021, 70, 159–170.
- Jia, J.; Gong, X.; Zhao, Y.; Yang, Z.; Ji, K.; Luan, T.; Zang, B.; Li, G. Autophagy enhancing contributes to the organ protective effect of alpha-lipoic acid in septic rats. Front. Immunol. 2019, 10, 1491.
- Prauchner, C.A. Oxidative stress in sepsis: Pathophysiological implications justifying antioxidant co-therapy. Burns 2017, 43, 471–485.
- Gibellini, L.; De Biasi, S.; Paolini, A.; Borella, R.; Boraldi, F.; Mattioli, M.; Lo Tartaro, D.; Fidanza, L.; Caro-Maldonado, A.; Meschiari, M.; et al. Altered bioenergetics and mitochondrial dysfunction of monocytes in patients with COVID-19 pneumonia. EMBO Mol. Med. 2020, 12, e13001.
- McLain, A.L.; Szweda, P.A.; Szweda, L.I. α-Ketoglutarate dehydrogenase: A mitochondrial redox sensor. Free Radic. Res. 2011, 45, 29–36.
- Tretter, L.; Adam-Vizi, V. Alpha-ketoglutarate dehydrogenase: A target and generator of oxidative stress. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 2335–2345.
- Guloyan, V.; Oganesian, B.; Baghdasaryan, N.; Yeh, C.; Singh, M.; Guilford, F.; Ting, Y.S.; Venketaraman, V. Glutathione supplementation as an adjunctive therapy in COVID-19. Antioxidants 2020, 9, 914.
- Pfaff, A.R.; Beltz, J.; King, E.; Ercal, N. Medicinal Thiols: Current Status and New Perspectives. Mini-Rev. Med. Chem. 2019, 20, 513–529.
- Hinson, J.A.; Roberts, D.W.; James, L.P. Mechanisms of acetaminophen-induced liver necrosis. Handb. Exp. Pharmacol. 2010, 196, 369–405.
- Elshazly, S.M.; El-Moselhy, M.A.; Barakat, W. Insights in the mechanism underlying the protective effect of α-lipoic acid against acetaminophen-hepatotoxicity. Eur. J. Pharmacol. 2014, 726, 116–123.
- Mahmoud, Y.I.; Mahmoud, A.A.; Nassar, G. Alpha-lipoic acid treatment of acetaminophen-induced rat liver damage. Biotech. Histochem. 2015, 90, 594–600.


