The nuclear receptor superfamily comprises a large group of proteins with functions essential for cell signaling, survival, and proliferation. There are multiple distinctions between nuclear superfamily classes defined by hallmark differences in function, ligand binding, tissue specificity, and DNA binding. In this review, we utilize the initial classification system, which defines subfamilies based on structure and functional difference. The defining feature of the nuclear receptor superfamily is that these proteins function as transcription factors. The loss of transcriptional regulation or gain of functioning of these receptors is a hallmark in numerous diseases. For example, in prostate cancer, the androgen receptor is a primary target for current prostate cancer therapies. Targeted cancer therapies for nuclear hormone receptors have been more feasible to develop than others due to the ligand availability and cell permeability of hormones.
1. Introduction
The nuclear receptor superfamily is a family of transcription factors that are widely expressed throughout the body. This family functions in well-organized signaling pathways that heavily rely on tissue microenvironment and when disrupted, endogenously or exogenously, can cause organ dysfunction, cancer, or loss of tissue integrity. Pharmacological intervention inhibiting signaling pathways of members of this family has been used for treatment of many diseases. Based on the evolution and robust treatment response to anti-androgen therapies, we examine different agents currently used in different stages of prostate cancer progression as well as new targets being explored due to a rise in treatment resistance.
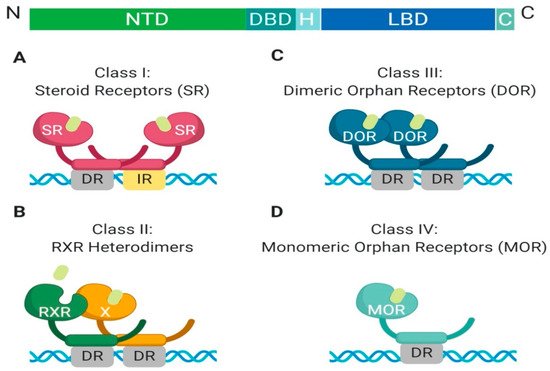
The nuclear receptor superfamily is comprised of over 500 members. This superfamily is further divided into four classes based on key characteristics such as dimerization, DNA binding motifs and specificity, and ligand binding. The four classes include steroid Receptors (Class I), RXR heterodimers (Class II), homodimeric orphan receptors (Class III), and monomeric orphan receptors (Class IV). Although there are some significant structural and functional differences between the classes, some key structural components are preserved, which are permissive to their respective functions (
Figure 1)
[1].
Figure 1. Schematic illustration of the classical nuclear receptor superfamily. (A–D) graphically represent the four classes of the nuclear receptor superfamily which are defined based on dimerization (homo, hetero, or mono), DNA binding (direct repeat or inverted repeat), and ligand specificity (required, or not required). Class I, Steroid Receptor (also known as nuclear hormone receptors); Class II, RXR Heterodimers; Class III, Dimeric Orphan Receptors; Class IV, Monomeric Orphan Receptors. Abbreviations: NTD, N-terminal domain; DBD, DNA-binding domain; H, Hinge region; LBD, Ligand-binding domain; C, Variable C-terminus; DR, Direct Repeat; IR, Inverted Repeat.
All nuclear receptor superfamily members contain a variable N-terminal domain (NTD), a DNA binding domain (DBD), a hinge region, a conserved ligand-binding domain (LBD), and a variable C-terminal domain. The two most highly conserved domains amongst all nuclear receptors are the DNA binding domain and the ligand-binding domain. The DNA binding domain contains two zinc finger motifs, which act as a hook, that allow binding to chromatin within the nucleus
[2]. Each class has different DNA binding recognition sequences, which range from variable half-sites with inverted repeats, direct repeats, or no repeats within the DNA sequence
[1].
The ligand-binding domain of nuclear receptors remains highly conserved in function but differs in specificity and affinity to specific ligands
[1][3]. All classes, excluding orphan receptors, are ligand-activated. Ligand binding at the LBD induces an allosteric change, inducing activation
[1][3]. Ligands within each class of nuclear receptors have similar structures. Furthermore, classification of the ligand determines which class of nuclear receptors each belongs to
[1][3]. For example, endogenously expressed ligands for these receptors can be hormones, metabolites, or enzymatic ligands, as well as unidentified ligands
[1][3].
Another feature which differentiates class members is partner dimerization within the nucleus. Classes I–III require dimerization while Class IV does not. Additionally, Class I and III require homodimerization, which can provide stronger zinc finger binding to DNA, while Class II requires heterodimerization
[1].
There have been modifications to each subclass based on new information gathered through structural analysis and sequencing data. For this review we will focus on the classical subdivisions of the nuclear receptor superfamily defined by the hallmarks of nuclear receptor superfamily structure and function such as dimerization, DNA binding motifs and specificity, and ligand-binding activation.
2. Class I: Overview of Steroid Hormone Receptors, Structure and Function
All members of Class I are grouped based on shared characteristics and functions (
Figure 2). First, they are ligand-activated receptors, for ligand-binding induces a conformational change that allows for homodimerization and subsequent DNA-binding. Additionally, Class I members have a unique role in the maintenance of cellular homeostasis, gene expression regulation in embryogenesis, and tissue development, as well as their ability to respond to extracellular signals in an endocrine manner, which allows the cells to adapt to systemic environmental changes. Within Class I nuclear hormone receptors, there can be redundancy of individual members to perform each other’s transcriptional functions, but it is highly dependent on tissue-specific expression of endogenous ligands
[4].
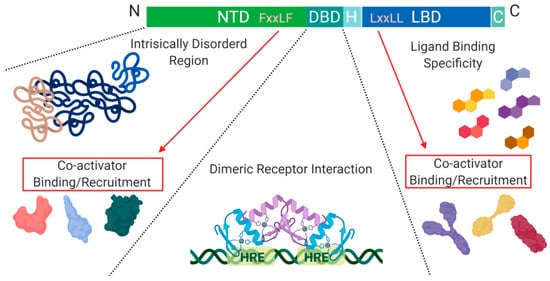
Figure 2. Schematic illustration of the nuclear receptor hormone family. Structural differences within the nuclear receptor hormone family occur at the NTD, DBD, and LBD. The functional differences are defined based on co-activator recruitment, dimeric receptor interactions, and ligand binding. Abbreviations: NTD, N-terminal domain; DBD, DNA-binding domain; H, Hinge region; LBD, Ligand-binding domain; C, Variable C-terminus; HRE, Hormone Response Elements.
Another level of regulation, which has been best characterized for Class I nuclear receptors, is determined by the presence of co-activators and/or repressors within the nucleus. These can be specific to a particular receptor within the class and are critical for transcription initiation or repression
[4].
The DNA binding domain (DBD), is a cysteine-rich domain that has a conserved amino acid sequence and encodes two zinc (Zn) finger motifs. Specifically, C1 to C4 are responsible for the first zinc finger motif, and C5 to C8 are responsible for forming the second. Each finger motif then chelates a Zn(II) ion, allowing for a structural DNA recognition site to form
[2]. The zinc finger motifs are known as the P-box and D-box, where the P-box refers to the 1st zinc finger motif in the sequence, which directly interacts with DNA, while the 2nd zinc finger site does not
[5]. The half site sequences on DNA that allow for zinc finger binding are highly conserved, as seen in the estrogen and glucocorticoid receptors. This level of similarity leads to single nucleotide or amino acid mutations of a zinc finger domain to cause receptor protein promiscuity. Wherein, receptor proteins can recognize hormone response elements of other receptors on DNA, and initiate transcription of genes non-specific to the external signal received
[2]. Additionally, the hexameric half-sites recognized by zinc finger motifs for the androgen receptor, progesterone receptor, and glucocorticoid receptor are highly conserved
[5]. However, the specific difference that allows for response element specificity is how the zinc fingers interact with each other once a dimer is formed, either head-to-head or head-to-tail
[6].
Another conserved structural feature among Class I nuclear receptors is their ligand-binding domain (LBD). The LBD contains around 12 α-helices, three of which form the hydrophobic pocket, also known as the ligand-binding pocket (LBP). The ligand binding specificity within the pocket is determined by conformational differences which cause steric hindrance of non-specific ligands
[7].
A structural feature of nuclear hormone receptors that has previously been overlooked is the activation function-1 (AF-1) protein domain within the N-terminal region and the activation function-2 (AF-2) in the LBD. This is a common feature found in Class I–III, but not in Class IV
[8]. The presence of AF-1 in the intrinsically disordered region of the N-terminal allows for flexibility and becomes ordered when bound to individual partners
[8]. On the C-terminal end, AF-2 requires ligand-binding to become active but remains ordered in all states
[8]. Unfortunately, when AF-2 is spliced out, the protein can undergo a gain of function mutations that no longer require ligand-binding for activation and cause dysregulated protein expression
[8]. On the outside of the AF-2 protein domain, at α-helix 12, there is a hot spot for steroid co-activator binding (SRC) mediated through its LxxLL motif, which promotes transcriptional activity (
Figure 2)
[9]. Similarly, within the NTD, there is a five amino acid long motif FxxLF that binds and stabilizes the N-terminal and C-terminal domains. This binding promotes the stabilization of dimers through an active conformational state, preventing ligand-bound dissociation
[5][9][10].
The N-terminal and C-terminal of the nuclear hormone receptors are crucial for the recruitment of co-activators within the nucleus, which can vary significantly between family members
[11]. The variability between nuclear hormone receptor co-activator binding is most likely caused by the specific amino acid arrangement in the NTD rather than difference in the chemical characteristics of amino acids present
[12]. Binding of the NTD with its preferred co-activator results in a highly coiled structure, which can alter the structural properties of the receptor. For example, the androgen receptor, in this highly coiled-state, becomes highly resistant to proteases
[12].
Overall, nuclear hormone receptors play a crucial role in body homeostasis. Thus, mutations, misfolding, or alteration of signaling pathways can often lead to systemic organ dysfunction. Each nuclear hormone receptor has a specific set of target genes, which display tissue-specificity, under a ligand-activated state initiated by a specific ligand.
2.1. Class I: The Androgen Receptor, Structural and Functional Differences
The androgen receptor (AR) is essential for male sexual differentiation, bone growth, muscle homeostasis, and development
[5]. AR is activated when α-dihydrotestosterone (DHT) binds to the LBP within the LBD of AR, inducing a conformational change. This leads to the activation of AR through the disassembly of chaperone proteins such as HSP70 and HSP90 and simultaneous exposure of a nuclear localization signal (NLS) in the DBD
[5][13].
Unlike other nuclear hormone receptors, androgen response elements have high specificity and low-affinity interactions with DNA
[5]. The literature suggests AR requires increased stability to bind DNA at specific androgen response element sites through head-to-head zinc finger dimerization
[5]. Additionally, AR has increased specificity to DNA recognition sites by recognizing both an inverted repeat and a direct repeat known as ADR3
[6]. Comparatively, the glucocorticoid receptor (GR) has been shown to have less bulky amino acids in the zinc finger motifs which form an open pocket within the head-to-head zinc dimer
[6]. The AR contains amino acids that allow for a more compact structural conformation which reduces pocket size, ultimately increases homodimer stability
[5][6][12].
The NTD of AR fosters a plethora of protein–protein interactions due to the variability in poly-glutamine and poly-glycine length which contributes to its highly disordered nature
[5][12]. The variability in glycine and glutamine residue repeats in the NTD of AR allow for interaction with numerous binding partners due to increased flexibility, increased number of conformations, and modified functionality
[5][14]. A decreased amount of glutamine and glycine repeats increases the transcriptional activity of AR most likely due to decreased protein–protein interactions with co-repressor binding partners
[5][12].
Upregulation of AR splice variants is commonly observed in different malignancies:
ARV7 is a splice variant of AR commonly upregulated after androgen deprivation therapy (
Figure 3). ARV7 lacks an LBD and does not require a ligand for active transcription. Recent studies have shown that ARV7 can homodimerize with full length-AR and repress the transcription of tumor suppressor genes
[15]. The ARV7 splice variant is constitutively active and requires full length-AR to repress transcription
[15].
Figure 3. Schematic illustration of androgen receptor splice variants. The most common AR splice variants implicated in prostate cancer treatment resistance are ARV7 and ARV567es. Splice Variant ARV7 lacks the LBD and therefore does not require ligand binding for activation which allows for constitutive activity in the context of low ligand availability during PCa treatments; ARV7 can also bind AR-full length in the nucleus, promoting continuous transcriptional activity. Splice variant ARV567es has an exon skipping mutation for exons 567 but retains exon 8 and therefore can bind DNA and remain constitutively active without ligand activation. However, the remaining exon 8 can potentially allow for co-activator recruitment and various conformational differences independent of AR-full length and ARV7. Abbreviations: AR, Androgen receptor; NTD, N-terminal domain; DBD, DNA-binding domain; H, Hinge region; LBD, Ligand-binding domain; C, Variable C-terminus.
ARv567es is a splice variant of AR that lacks exons 5, 6, and 7 while retaining exon 8 and does not require ligand binding for transcriptional activity (
Figure 3)
[16]. The splice variant ARV567es requires homodimerization with full length-AR for actively transcribing target genes. Unique from other forms of AR, this splice variant localizes to the nucleus wherein it waits for full length-AR to begin any activity
[16]. Recent studies identify that ARv567es actively transcribes a unique set of target genes that are distinct from full length-AR, indicating that expression of ARv567es could be a fail-safe mechanism used by tumor cells to promote cell survival
[16].
2.2. Class I: The Progesterone Receptor, Structural and Functional Differences
The ligand for the progesterone receptor (PR) is progesterone. Ligand-activation of PR plays a critical role in female mammary gland development/homeostasis and other female reproductive organs
[10]. Additionally, PR is expressed in prostate tissue and plays an essential role in tissue microenvironment and stromal cell differentiation
[17]. PR has two isoforms which have differential roles in normal organ functioning and the balanced expression of both isoforms is critical for normal tissue function
[10][18]. The two isoforms—Progesterone Receptor-A (PR-A) and Progesterone Receptor-B (PR-B)—differ in size at the NTD (
Figure 4)
[10].
Figure 4. Schematic illustration of progesterone receptor isoforms. The progesterone receptor has two distinct isoforms, PR-A and PR-B. PR-B is more transcriptionally active due to the extended N-terminal domain that contains AF3 (unique domain), an activation function unit that allows for an increase in co-binding partners and activity. The isoform PR-A is less transcriptionally active than PR-B and primarily functions in non-genomic pathway activity. Its upregulation is implicated in cancers. Abbreviations: PR, Progesterone receptor; NTD, N-terminal domain; DBD, DNA-binding domain; H, Hinge region; LBD, Ligand-binding domain; C, Variable C-terminus; AF1, Activation function-1; AF2, Activation function-2; AF3, Activation function-3.
PR-B has an extended NTD, which is called a unique domain or activating function domain 3 (AF-3)
[10]. PR-B is more transcriptionally active than PR-A and plays a crucial role in mammary development. However, dominant expression of PR-A is implicated in cancer onset due to non-genomic activities mediated through the binding of activated Src kinases
[10].
Post-translational modifications of the hinge region of PR-A/B allows the PR to interact with chromatin-associated high mobility proteins 1/2 (HMGB1/2) and Jun dimerization protein 2 (Jun2)
[19][20]. These interactions have been shown to increase transcriptional activity of PR
[20]. HMGB1/2 is a protein that increases DNA-protein binding interactions indirectly through increasing the number of contacts of the PR to DNA through dynamic conformational change
[21][22][23].
Furthermore, recent evidence suggests a role of PR in prostate cancer progression, specifically through its role in regulating smooth actin muscle (SMAα) in stromal cells
[17][24]. PR levels were upregulated after castration in patients with prostate cancer, which could contribute to treatment resistance due to its role in stromal cell differentiation and mobility
[17][24]. Until recently, the role of PR in prostate cancer was not evaluated but maybe a key player in understanding current mechanisms of treatment resistance.
2.3. Class I: The Estrogen Receptor, Structural and Functional Differences
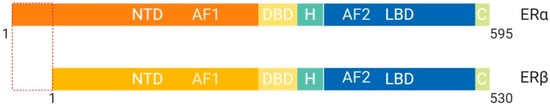
The estrogen receptor (ER), whose ligand is estradiol, plays a key role in female reproduction
[18]. There are two isoforms of the ER known as ERα and ERβ, which are structurally distinct and perform different functions
[18] (
Figure 5).
Figure 5. Schematic illustration of estrogen receptor isoforms. The estrogen receptor has two isoforms, ERα and ERβ, and is comparatively smaller than other family members. The isoform ERα has an extended NTD which allows for more transcriptional activation, whereas, ERβ has a shorter NTD and is less transcriptionally active. Most often the ERα isoform is upregulated in cancers. Abbreviations: ER, Estrogen receptor; NTD, N-terminal domain; DBD, DNA-binding domain; H, Hinge region; LBD, Ligand-binding domain; C, Variable C-terminus; AF1, Activation function-1; AF2, Activation function-2.
These structural differences allow for interaction with different binding partners and subsequent transcriptional activation of distinct target genes
[4]. ERβ protein lacks an AF-1 region, which suggests that ligand binding is essential for function through LxxLL motif binding at AF-2 α-helix 12
[4]. ERα protein contains both AF-1 and AF-2 structural features and is more transcriptionally active than ERβ
[4]. Previous studies have shown that the ER binds multiple estrogen response elements (ERE) that vary based on the second didactic half-site
[4]. It is suggested that the variability of ERE allows for modulation of allosteric regulation and ultimately, co-activator recruitment
[4]. Post-translational modifications of ERα such as methylation of Arg
260 by protein arginine methyltransferase (PMT1) are necessary for ERα to interact with the p85 subunit of PI3K and c-SRC
[25]. On the other hand, acetylation of Lys
266/268 by p300 enhances the transcriptional activity of ERα by increasing ERE binding specificity
[26].
Recent evidence suggests that ERβ plays an essential role in the proliferation of prostatic epithelial cells, which is a feature of prostate cancer progression
[27]. The downregulation of ERβ as a consequence of current prostate cancer treatments could increase the proliferation of prostatic epithelial cells and contribute to disease progression. Furthermore, studies show that a subset of patients with high-grade tumors had a loss of the ERβ gene locus
[27]. Similarly, mice that had a loss of ERβ developed prostate cancer, which could be a useful biomarker for early detection of prostate cancer
[27].
ER has a pivotal role in the female reproductive system and secondary sexual characteristic development and function
[4][28]. ER has been well characterized as a key player in breast cancer development. The differential expression of ER’s isoforms has been implicated in breast cancer metastasis, and can be used to determine treatment, prognosis, and stage of the disease
[29]. Similar to prostate cancer, some forms of breast cancer are also hormone-sensitive, with approximately 70% of them being hormone-sensitive and ER positive
[30]. Based on the currently available targeted therapies for breast cancer (also referred to as Endocrine Therapy (ET)) and their superiority to chemotherapy with regards to tolerance, efficacy, and less severe side effects, breast cancer is subdivided into distinct biologic groups based on receptor expression: Estrogen Receptor (ER+), Progesterone Receptor (PR+), those that express the epidermal growth factor receptor 2 (HER2+), and those that do not express either are classified as triple negative BC
[30].
Production of estrogen in females is analogous to testosterone production in males. This allows for some of the same agents used for chemical castration to be used for ovarian ablation (see below). The use of LHRH analogs allows for the downregulation of estrogen production by the ovaries, the main source of estrogen in pre-menopausal women. Aromatase inhibitors (i.e., anastrozole, exemestane and letrozole) inhibit the enzyme aromatase, which converts androgens into estrogens in tissues outside of the ovaries. This therapy works best in post-menopausal women, since production of estrogen post-menopause is not in the ovaries
[30].
2.4. Class I: The Glucocorticoid Receptor, Structural and Functional Differences
The ligands for the glucocorticoid receptor (GR) are glucocorticoids which are produced by the adrenal cortex. This protein is ubiquitously expressed throughout the body and mediates metabolism as well as anti-inflammatory response through ligand-binding activation of cortisol. Recent studies have shown that specificity of DNA binding for GR is mediated by amino acid composition in the DBD of the protein which most likely contributes to the high level of protein expression throughout the entire body but lack of overlapping function with family members such as AR
[31]. More specifically, one amino acid polymorphism will allow for a different conformation and therefore lead to different target gene binding
[31]. However, mutation of the DNA binding domain could also decrease DNA binding specificity and allow for activation of target genes that belong to other family members such as AR
[31]. Furthermore, GR, unlike other nuclear hormone receptors, has an abundance of acidic residues in the NTD, which increase its interaction with co-activator proteins
[32]. Additionally, the AF-1 in the NTD of GR can perform 65% of normal functioning compared to wildtype GR
[32]. Most other nuclear receptor family members require both AF-1 and AF-2 for proper function. However, the modification of acidic residues in the NTD allows for GR to function in the absence of a ligand
[32].
More recent evidence suggests that TIF2.0 (p160 co-activator TIF2) directly interacts with the NTD of GR
[33][34]. Previously, TIF2 has been shown to solely interact with the LBD of other nuclear hormone receptors wherein, TIF2.0 has an extended NTD
[34]. Through NTD binding of TIF2.0 to GR, a conformational change occurs, allowing for an increased α-helix formation
[33]. Similarly, binding of TIF2.0-GR was shown to inhibit co-repressor binding, which suggests a unique mechanism of increased transcriptional activity of GR
[33].
3. Class II: RXR Heterodimers, Structure and Function
The Retinoid X Receptor (RXR) ligand is 9-cis-retinoic acid or alitretinoin, which plays a role in lipid metabolism, apoptosis, and the immune system
[35]. RXR unlike nuclear hormone receptors, are highly promiscuous with regards to their binding partners
[3]. The key feature of Class II nuclear receptors is that RXR dimerization is required for activation. The RXR can bind to itself and promote activation, but other members of this family such as Peroxisome Proliferator Activator Receptor (PPAR), Pregnane X Receptor, and Liver X Receptor all require heterodimerization with RXR to translocate to the nucleus
[3]. All receptors in Class II bind to unique response elements, which makes RXR dynamic and heavily relied upon for normal physiological function
[36]. Downregulation or loss of RXR signaling has been shown to promote inflammation of vital organ systems such as the liver
[37]. Interestingly, RXR without ligand binding can still bind DNA and perform functions such as recruiting co-repressor complexes to repress gene expression through heterodimerization with Retinoic Acid Receptor (RAR)
[38]. In cancer cells, RXR is sequestered in the cytoplasm by the co-repressor complex AEG-1/MTDH/LYRIC which decreases its transcriptional activity
[38].
4. Class III: Homodimeric Orphan Receptors, Structure and Function
Homodimeric orphan receptors are different from Class I and II in that no ligand has been identified for their activation
[1][3]. Homodimeric orphan receptors are structurally similar to other family members but differ in sequence-specific binding to DNA
[3]. The Class III nuclear receptor family bind to direct repeat and palindromic sequences
[3]. Additionally, homodimeric orphan receptors have highly constitutive transactivation and transrepression functions, suggesting that perhaps no ligand is required for activation
[3]. In some cases, Class III receptors bind similar target genes as nuclear hormone receptors and therefore, may play a critical role in alternative pathway activation
[1][3]. For example, RevERbAα is an example of a homodimeric, as well as a monomeric, orphan receptor. When RevErbAα binds as a dimer to DNA, it acts as a transcriptional repressor for genes that are typically activated by Class II RXR-RAR receptors
[39]. Interestingly, RORα is a Class IV, a monomeric orphan receptor that is critical for transcriptional activation of genes essential for proper cerebellar development
[40][41]. A gain of function mutation at the zinc finger binding motifs of RORα allows the receptor to bind as a dimer to a subset of target genes while still maintaining its original function. However, compared to the well-characterized Class I receptors, there is a lack of functional and structural information that differentiates Class III receptors
[41].
5. Class IV: Monomeric Orphan Receptors, Structure and Function
Monomeric orphan receptors are similar to Class III in that they do not require a ligand for activation. However, functionally they have a distinct role in steroid synthesis
[42]. The Class IV nuclear receptor, Steroidogenesis Factor-1 (SF-1) monomerically binds to steroidogenic enzymes at the DNA enhancer sequence in all tissues responsible for steroid synthesis
[3][43]. SF-1 is expressed in all steroidogenic tissues where the receptor remains constitutively active
[42]. It was shown that the loss of gene expression of SF-1 results in the failure of organ development during embryogenesis
[42][43]. While Class IV nuclear receptors are not directly involved in hormone signaling, this class plays a critical role throughout early sexual differentiation as well as in hormone biosynthesis
[43]. Similar to Class III nuclear receptors, there is still a lot to be understood regarding the structure and function of monomeric orphan receptors.
 +1 credit
+1 credit