 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hideki Tatsukawa | + 2146 word(s) | 2146 | 2021-07-27 12:01:00 | | | |
| 2 | Ron Wang | + 187 word(s) | 2333 | 2021-08-10 05:31:40 | | | | |
| 3 | Ron Wang | Meta information modification | 2333 | 2021-08-11 10:39:51 | | |
Video Upload Options
Transglutaminase 2 (TG2) is a ubiquitously expressed enzyme catalyzing the crosslinking between Gln and Lys residues and involved in various pathophysiological events. Besides this crosslinking activity, TG2 functions as a deamidase, GTPase, isopeptidase, adapter/scaffold, protein disulfide isomerase, and kinase. It also plays a role in the regulation of hypusination and serotonylation.
1. Introduction
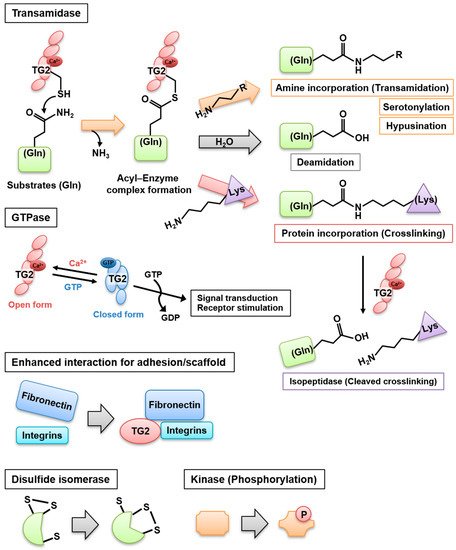
Transglutaminases (TGase) are multifunctional enzymes and constitute a family of eight isozymes designated as blood coagulation factor XIII and TG1–7. In this family, TG2 is widely distributed and involved in multiple biological processes. It catalyzes a Ca 2+ -dependent acyl transfer reaction between the γ-carboxamide group of glutamine present in a particular sequence and either primary amines, such as polyamines and histamine, or the ε-amino group of a lysine residue, intra- or inter-molecularly. Water replaces the amine donor substrates, leading to the deamidation of glutamine. In addition, TG2 and factor XIIIa exhibit a Ca 2+ -dependent isopeptidase activity and can hydrolyze the isopeptide bond, at least under test tube conditions. TG2 exerts additional enzymatic activities that do not require Ca 2 + , i.e., it hydrolyzes ATP and GTP to mediate signal transduction through G-protein-coupled receptors, protein disulfide isomerases, and protein kinases, as well as interacts with several proteins as an adhesion or scaffold protein [1] ( Figure 1 ).
TG2 is ubiquitously distributed inside (in the nucleus, cytoplasm, plasma membrane, and mitochondria) and outside the cell, where it appears in the extracellular matrix (ECM) and exosome [2][3][4]. In mammals, TG2 is detected across the body, including the blood, extracellular spaces, and intracellular compartments of nearly all tissues. It is involved in cell death, growth, and differentiation as well as tissue repair by tissue remodeling/wound healing and ECM assembly [5]. In this article, we focus on the role of TG2 in cell death, macrophage activation, and tissue repair processes, which are involved in several pathogeneses, including tissue injury, inflammation, and fibrosis. This review aims to summarize the recent knowledge on the mechanisms activated by TG2 to regulate cell death/survival and fibrosis in the tissue repair process.
2. Multifunctional Activity and Regulation of TG2
When cells are exposed to increased intracellular Ca 2+ concentrations (>700–800 nM) in response to certain stimuli, including injury and inflammation signals, TG2 structural conformation is dramatically altered and changes from a closed to an opened form that exerts crosslinking and transamidase activities [6][7]. In the appropriate redox condition, a TG2 intermediate thioester is formed through the attack of an acyl donor (γ-carboxamide group of a protein glutamine residue) by the nucleophilic active thiolate (cysteine residue at the active site of TG2), with release of ammonia. Then, the thiolate is restored via the nucleophilic attack of an acyl acceptor (ε-amino group of a protein lysine residue), leading to the formation of a covalent intra- or inter-molecular N ε -(γ-glutamyl)lysine isopeptide bond, which is resistant to degradation [8]. It has been suggested that the isopeptide bond contributes to the stabilization of the ECM and the prevention of the release of the intracellular content from apoptotic cells into the extracellular milieu. A similar reaction also occurs through the incorporation of primary amines and polyamines into the γ-carboxamide group of a protein glutamine residue.
If the aforementioned intermediate thioester bond is attacked by a water molecule as the acyl acceptor, a deamidation reaction occurs, in which the site-specific acyl-donor glutamine is converted to a glutamate residue [3]. For many years, the deamination reaction was believed to occur as a side reaction of the absence of primary amines or at low pH, when amine availability was limited. In these conditions, water would play a role owing to its abundance [9][10]. However, site-specific deamidations of heat shock protein [11] and βB2/3-crystallines [12] have been reported, suggesting that the substrate affinity for TG2 and the reaction conditions influence the propensity toward deamidation or transamidation [13].
The reversible crosslinking of α2-plasmin inhibitor to fibrinogen and fibrin by factor XIIIa was reported and is potentially involved in the regulation of fibrinolytic processes [14][15]. Biochemical studies demonstrated that TG2 also exhibits an isopeptidase activity targeting N ε -(γ-glutamyl)lysine [16]. Therefore, an unknown regulatory system of TG2 might exist to separately switch on or off the transamidase and isopeptidase activities. Specific TG2 mutants, which exhibit deficient transamidase (W332F) and isopeptidase (W278F) activities, have been identified [17]. Further research might help elucidate the role of the TG2 isopeptidase activity in physiological and pathological processes.
TG2 was reported to demonstrate a protein disulfide isomerase (PDI) activity, which has been implicated in mitochondrial-dependent apoptosis [18][19]. TG2 also exerts an intrinsic serine/threonine kinase activity to phosphorylate insulin-like growth factor binding protein-3 [20], p53 tumor suppressor protein [21], histones H1–4 [22], and retinoblastoma (Rb) protein [23]. Furthermore, TG2 affects hypusine metabolism, regulating the activity of eukaryotic initiation factor 5A and cell proliferation [24]. Recently, TG2 was reported to serotonylate histone H3 trimethylated lysine 4 (H3K4me3)-marked nucleosomes, controlling the recruitment of transcription factors, including TFIID [25].
3. Regulation of TG2 Expression and Activity
TGM2 gene expression is regulated by various cellular events, including apoptotic stimuli [26][27], viral infection [28], endoplasmic reticulum (ER) stress [29][30], hypoxia/ischemia [31][32][33], inflammation [34], tissue remodeling [35], and cancer [36][37][38]. It is mediated by several related factors and cytokines, such as retinoids [39][40], lipopolysaccharides [41], transforming growth factor (TGF)-β/bone morphogenetic protein 4 [42], nuclear factor-κ B (NF-κB) [38][43], glucocorticoids [44], interleukin (IL)-1 [45], IL-6 [46], hypoxia-inducible factor-1 [47], tumor necrosis factor (TNF)-α [48], and epidermal growth factor (EGF) [36]. Retinoic acid is a well-known inducer of TG2 expression and promotes the cellular differentiation of neutrophil granulocytes [49][50] and neuroblastoma cells [51][52] through the heterodimer retinoid acid receptor (RAR)/retinoid X receptor (RXR) and transcription factor Sp1 [40]. TG2 expression is upregulated in cancer cells resistant to chemotherapy or with high metastatic potential. TGM2 promoter contains response elements to inflammation and hypoxia, which are greatly elevated in the environment surrounding malignant tumors, leading to an increased expression of TG2 [47]. Ischemia also promotes TG2 expression [31]. In addition, n-Myc and c-Myc contribute to the regulation of TG2 expression by recruiting histone deacetylase 1 protein to the TGM2 promoter in cancer cells [53]. Interestingly, the antiproliferative effects of histone deacetylase inhibitors in cancer cells are impaired by the induction of TG2 mRNA and protein expression, suggesting that TG2 is involved in the resistance of cancer cells [54]. Since the half-life of TG2 is about 10 h in colorectal cancer HT29 cells, the sustained protein synthesis of TG2 is necessary for cancer proliferation and resistance to anticancer drugs such as histone deacetylase inhibitors [54]. TG2 influences TGF-β activation and signaling, whereas TGF-β1 was reported to promote and suppress TG2 expression [42][55]. TG2 expression is increased by several cytokines, such as IL-6, TNF-α, and NF-κB, in many cell types, including human hepatoblastoma cells [46][48] and macrophages [56][57]. AF4/FMR2 family member 1, known as a central scaffolding protein of super elongation complex, was recently reported to contribute to TG2 expression after being recruited to the TGM2 promoter in mouse adipocytes [58].
Human and murine TGM2 promoters are well characterized and contain various response elements for retinoic acid (−1731 bp and −1720 bp), glucocorticoids (−1399 bp), NF-κB (−1338 bp), IL-6 (−1190 bp) , TGF-β1 (−900 bp), estrogens (−656 bp) [59][60], activator protein-2 (AP-2, −634 bp), AP-1 (−183 bp), hypoxia (−367 bp), and nuclear factor-1 (+4 bp, +12 bp) [40][61], as well as motif regions, such as CAAT box (−96 bp), TATA box (−29 bp), and GC box, for Sp1 binding (−54 bp, −43 bp, +59 bp, +65 bp) [62]. Figure 2 A presents the regulatory elements previously reported for the human TGM2 promoter. In addition, TG2 expression is directly downregulated by micro-RNA 19, which is responsible for the increased invasion and metastasis of colorectal cancer cells [63]. In addition, enhancer RNA molecules of TG2 expression were identified to regulate the recruitment of the transcriptional repressor CTCF in the intergenic region of thymocytes treated with retinoids and TGF-β to induce their death [64]. Furthermore, a recent review summarized the various potential binding sites of transcription factors and single-nucleotide polymorphisms of the TGM2 promoter using information obtained from the public database of chromatin immunoprecipitation sequencing [65]. Finally, several splicing variants of TGM2 exhibit different regulatory properties and catalytic activities, affecting the global TG2 activity [66]. Therefore, TGM2 gene expression is regulated by multiple signaling pathways involved in physiological and pathological events, although these functional roles have not been fully elucidated.
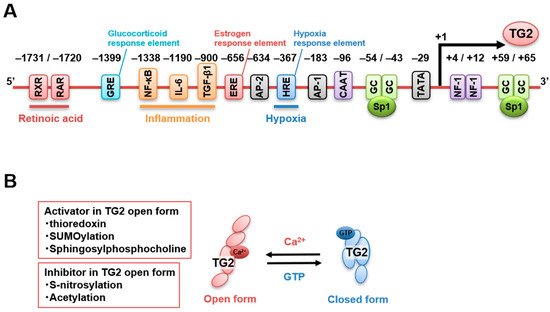
A few studies have focused on the regulation of TG2 activity compared with the number of investigations on its transcriptional regulation. Ca 2+ and GTP are known as a competitive activator and a suppressor of TG2 transamidase activity, respectively. Ca 2+ binding alters TG2 structural conformation by moving the β-barrel domains 3 and 4 away from the catalytic domain 2, opening the active center and facilitating access to the substrate. TG2 appears to be inactive in cells in the absence of stress (~100 nM free cytoplasmic [Ca 2+ ]). In addition, a free cytoplasmic GTP concentration higher than 100 μM is required to maintain the closed GTP-bound conformation of TG2, which inhibits its transamidase activity [67].
The conditions in the extracellular space are suitable for TG2 activation as Ca 2 + and GTP are present at high and low levels, respectively. However, a highly oxidative state was reported to keep TG2 in the inactive state in the absence of stress due to the formation of disulfide bonds as a posttranslational modification [68], which was reversed by thioredoxin-mediated reduction [69]. Furthermore, extracellular TG2 can be negatively regulated by S-nitrosylation, indicating that nitric oxide is also a potent inhibitor of TG2 activation and might be involved in age-related vascular stiffness [70][71]. TG2 acetylation was also reported to suppress its activity in vitro [72]. Finally, TG2 is stabilized by SUMOylation, which inhibits TG2 ubiquitination, leading to enhanced protein levels and activity [73][74].
4. TG2 Functions in Fibrosis
The TG2 transamidase activity contributes to the wound healing process and fibrosis. The reaction products form an N ε -(γ-glutamyl)lysine isopeptide bond resulting from the crosslinking. This is an important step for the maturation and stabilization of ECM components, such as collagens, exacerbating scarring and fibrosis in various tissues, including the liver [75][76][77][78], kidney [79][80][81][82][83][84], lung [85][86][87][88][89][90], and heart [91][92]. The other enzymatic crosslinker, lysyl oxidase (LOX), has also been reported to contribute to collagen stabilization. LOX oxidizes certain lysine residues in collagen to produce aldehydes, which react to form covalent bonds and stabilize molecules within the collagen fibers [93]. Impaired crosslinking by LOX results in weak collagen fibers and fragile collagenous tissue [94]. In the remodeling of fibroblast-populated collagen lattices, TG2 predominantly contributes to the Ca 2+ -dependent early entrenchment (initial remodeling) by crosslinking of the extant matrix, whereas LOX implicates Ca 2+ -independent contractility at later times [95]. These results suggest that, in fibrosis, TG2 is involved in early ECM remodeling, while LOX contributes to subsequent modification.
Aside from the direct ECM stabilization, the TG2 transamidase activity appears to play a significant role in the fixation and activation of the profibrotic cytokine TGF-β. TGF-β is released in a latent form and converted to an active one. Enhanced TG2 activity is required for TGF-β activation from the latency binding complex as it promotes crosslinking of the large latent TGF-β binding protein to fibronectin or other ECM components on the cell surface [90][96][97][98][99]. The secretion of TG2 into the ECM is important for its function in TGF-β activation. The secretion mechanisms are unclear, as TG2 lacks the signal peptide necessary for ER targeting and the classical protein secretion mechanism through the ER–Golgi system. Moreover, no Golgi-associated protein modification, such as glycosylation, has been evidenced for TG2 [100]. Recent studies demonstrated that TG2 interacts with the heparan sulfate chains of proteoglycans, forms a complex with fibronectin, and interacts with integrins and heparan sulfate proteoglycans in the ECM to promote cell adhesion and spreading [101][102]. The interaction between TG2 and the heparan sulfate chains of cell surface syndecans is a potential mechanism implicated in the pathophysiological role of TG2, including in fibrosis [103][104][105].
As previously described, we demonstrated that nuclear TG2 inactivated Sp1 by crosslinking, leading to reduced expression of c-Met and consequently activation of hepatic apoptosis in a hepatic injury mouse model and in patients with alcoholic steatohepatitis [106]. TG2-mediated reduction of c-Met expression might be involved in the impaired hepatocyte regeneration observed in patients with alcoholic liver diseases [107][108][109]. Furthermore, hepatocyte-specific c-Met-deficient mice demonstrated more extensive liver cell damages and fibrosis, indicating that the induction of nuclear TG2/crosslinked Sp1 /downregulated c-Met axis accompanied liver fibrosis. In agreement with our findings, TG2 nuclear accumulation and crosslinked Sp1 were observed in the fibrotic area of patients with alcoholic steatohepatitis [110].
However, TG2-deficient mice demonstrated no alteration of the fibrosis levels in the liver after treatment with CCl4 or thioacetamide [111]. This contradictory result might be due to discrepancies in the method used to target the TGM2 gene, in the mouse background, and in the disease model. We obtained similar results using TG2-deficient mice. Indeed, liver fibrosis induced by bile duct ligation was not inhibited in these mice [77], although these mice presented a significant reduction of fibrosis induction in other fibrosis models, such as kidney fibrosis induced by unilateral ureteral obstruction [83] and lung fibrosis resulting from bleomycin treatment [87]. Interestingly, TG2 or pan-TGase inhibitors, including competitive or reversible/irreversible inhibitors, have been demonstrated to be consistently protective in several fibrosis models, including liver fibrosis induced by both CCl4 and bile duct ligation [112][77][83][113][114][115][116][117][118].
References
- Hitomi, K.; Kojima, S.; Fesus, L. Transglutaminases; Hitomi, K., Kojima, S., Fesus, L., Eds.; Springer: Tokyo, Japan, 2015; ISBN 978-4-431-55823-1.
- D’Eletto, M.; Rossin, F.; Fedorova, O.; Farrace, M.G.; Piacentini, M. Transglutaminase type 2 in the regulation of proteostasis. Biol. Chem. 2019, 400, 125–140.
- Eckert, R.L.; Kaartinen, M.T.; Nurminskaya, M.; Belkin, A.M.; Colak, G.; Johnson, G.V.W.; Mehta, K. Transglutaminase regulation of cell function. Physiol. Rev. 2014, 94, 383–417.
- Iismaa, S.E.; Mearns, B.M.; Lorand, L.; Graham, R.M. Transglutaminases and disease: Lessons from genetically engineered mouse models and inherited disorders. Physiol. Rev. 2009, 89, 991–1023.
- Kanchan, K.; Fuxreiter, M.; Fésüs, L. Physiological, pathological, and structural implications of non-enzymatic protein-protein interactions of the multifunctional human transglutaminase 2. Cell. Mol. Life Sci. 2015, 72, 3009–3035.
- Begg, G.E.; Carrington, L.; Stokes, P.H.; Matthews, J.M.; Wouters, M.A.; Husain, A.; Lorand, L.; Iismaa, S.E.; Graham, R.M. Mechanism of allosteric regulation of transglutaminase 2 by GTP. Proc. Natl. Acad. Sci. USA 2006, 103, 19683–19688.
- Savoca, M.; Tonoli, E.; Atobatele, A.; Verderio, E. Biocatalysis by transglutaminases: A review of biotechnological applications. Micromachines 2018, 9, 562.
- Keillor, J.W.; Clouthier, C.M.; Apperley, K.Y.P.; Akbar, A.; Mulani, A. Acyl transfer mechanisms of tissue transglutaminase. Bioorg. Chem. 2014, 57, 186–197.
- Fleckenstein, B.; Molberg, Ø.; Qiao, S.-W.; Schmid, D.G.; von der Mülbe, F.; Elgstøen, K.; Jung, G.; Sollid, L.M. Gliadin T cell epitope selection by tissue transglutaminase in celiac disease. Role of enzyme specificity and pH influence on the transamidation versus deamidation reactions. J. Biol. Chem. 2002.
- Sivadó, É.; El Alaoui, M.; Kiraly, R.; Fesüs, L.; Delolme, F.; Page, A.; El Alaoui, S. Optimised methods (SDS/PAGE and LC-MS) reveal deamidation in all examined transglutaminase-mediated reactions. FEBS Open Bio 2019, 9, 396–404.
- Boros, S.; Åhrman, E.; Wunderink, L.; Kamps, B.; De Jong, W.W.; Boelens, W.C.; Emmanuelsson, C.S. Site-specific transamidation and deamidation of the small heat-shock protein Hsp20 by tissue transglutaminase. Proteins Struct. Funct. Genet. 2006, 62, 1044–1052.
- Boros, S.; Wilmarth, P.A.; Kamps, B.; de Jong, W.W.; Bloemendal, H.; Lampi, K.; Boelens, W.C. Tissue transglutaminase catalyzes the deamidation of glutamines in lens βB2- and βB3-crystallins. Exp. Eye Res. 2008, 86, 383–393.
- Stamnaes, J.; Fleckenstein, B.; Sollid, L.M. The propensity for deamidation and transamidation of peptides by transglutaminase 2 is dependent on substrate affinity and reaction conditions. Biochim. Biophys. Acta Proteins Proteom. 2008, 1784, 1804–1811.
- Ichinose, A.; Aoki, N. Reversible cross-linking of α2-plasmin inhibitor to fibrinogen by fibrin-stabilizing factor. Biochim. Biophys. Acta Protein Struct. Mol. 1982, 706, 158–164.
- Mimuro, J.; Kimura, S.; Aoki, N. Release of α2-plasmin inhibitor from plasma fibrin clots by activated coagulation factor XIII. Its effect on fibrinolysis. J. Clin. Investig. 1986, 77, 1006–1013.
- Parameswaran, K.N.; Cheng, X.F.; Chen, E.C.; Velasco, P.T.; Wilson, J.H.; Lorand, L. Hydrolysis of γ:ε isopeptides by cytosolic transglutaminases and by coagulation factor XIII(a). J. Biol. Chem. 1997, 272, 10311–10317.
- Király, R.; Thangaraju, K.; Nagy, Z.; Collighan, R.; Nemes, Z.; Griffin, M.; Fésüs, L. Isopeptidase activity of human transglutaminase 2: Disconnection from transamidation and characterization by kinetic parameters. Amino Acids 2016, 48, 31–40.
- Hasegawa, G.; Suwa, M.; Ichikawa, Y.; Ohtsuka, T.; Kumagai, S.; Kikuchi, M.; Sato, Y.; Saito, Y. A novel function of tissue-type transglutaminase: Protein disulphide isomerase. Biochem. J. 2003, 373, 793–803.
- Malorni, W.; Farrace, M.G.; Matarrese, P.; Tinari, A.; Ciarlo, L.; Mousavi-Shafaei, P.; D’Eletto, M.; Di Giacomo, G.; Melino, G.; Palmieri, L.; et al. The adenine nucleotide translocator 1 acts as a type 2 transglutaminase substrate: Implications for mitochondrial-dependent apoptosis. Cell Death Differ. 2009, 16, 1480–1492.
- Mishra, S.; Murphy, L.J. Tissue transglutaminase has intrinsic kinase activity. Identification of transglutaminase 2 as an insulin-like growth factor-binding protein-3 kinase. J. Biol. Chem. 2004, 279, 23863–23868.
- Mishra, S.; Murphy, L.J. The p53 oncoprotein is a substrate for tissue transglutaminase kinase activity. Biochem. Biophys. Res. Commun. 2006, 339, 726–730.
- Mishra, S.; Saleh, A.; Espino, P.S.; Davie, J.R.; Murphy, L.J. Phosphorylation of histones by tissue transglutaminase. J. Biol. Chem. 2006, 281, 5532–5538.
- Mishra, S.; Melino, G.; Murphy, L.J. Transglutaminase 2 kinase activity facilitates protein kinase A-induced phosphorylation of retinoblastoma protein. J. Biol. Chem. 2007, 282, 18108–18115.
- Beninati, S.; Gentile, V.; Caraglia, M.; Lentini, A.; Tagliaferri, P.; Abbruzzese, A. Tissue transglutaminase expression affects hypusine metabolism in BALB/c 3T3 cells. FEBS Lett. 1998, 437, 34–38.
- Farrelly, L.A.; Thompson, R.E.; Zhao, S.; Lepack, A.E.; Lyu, Y.; Bhanu, N.V.; Zhang, B.; Loh, Y.H.E.; Ramakrishnan, A.; Vadodaria, K.C.; et al. Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 2019, 567, 535–539.
- Rodolfo, C.; Mormone, E.; Matarrese, P.; Ciccosanti, F.; Farrace, M.G.; Garofano, E.; Piredda, L.; Fimia, G.M.; Malorni, W.; Piacentini, M. Tissue transglutaminase is a multifunctional BH3-only protein. J. Biol. Chem. 2004, 279, 54783–54792.
- Szegezdi, É.; Szondy, Z.; Nagy, L.; Nemes, Z.; Friis, R.R.; Davies, P.J.A.; Fésüs, L. Apoptosis-linked in vivo regulation of the tissue transglutaminase gene promoter. Cell Death Differ. 2000, 7, 1225–1233.
- Amendola, A.; Rodolfo, C.; Caro, A.; Ciccosanti, F.; Falasca, L.; Piacentini, M. “Tissue” Transglutaminase Expression in HIV-Infected Cells. Ann. N. Y. Acad. Sci. 2001, 946, 108–120.
- Kuo, T.F.; Tatsukawa, H.; Matsuura, T.; Nagatsuma, K.; Hirose, S.; Kojima, S. Free fatty acids induce transglutaminase 2-dependent apoptosis in hepatocytes via ER stress-stimulated PERK pathways. J. Cell. Physiol. 2012, 227, 1130–1137.
- Currò, M.; Condello, S.; Caccamo, D.; Ferlazzo, N.; Parisi, G.; Ientile, R. Homocysteine-induced toxicity increases TG2 expression in Neuro2a cells. Amino Acids 2009, 36, 725–730.
- Filiano, A.J.; Tucholski, J.; Dolan, P.J.; Colak, G.; Johnson, G.V.W. Transglutaminase 2 protects against ischemic stroke. Neurobiol. Dis. 2010, 39, 334–343.
- Tolentino, P.J.; Waghray, A.; Wang, K.K.W.; Hayes, R.L. Increased expression of tissue-type transglutaminase following middle cerebral artery occlusion in rats. J. Neurochem. 2004, 89, 1301–1307.
- Currò, M.; Ferlazzo, N.; Giunta, M.L.; Montalto, A.S.; Russo, T.; Arena, S.; Impellizzeri, P.; Caccamo, D.; Romeo, C.; Ientile, R. Hypoxia-dependent expression of TG2 isoforms in neuroblastoma cells as consequence of different MYCN amplification status. Int. J. Mol. Sci. 2020, 21, 1364.
- Liu, C.; Kellems, R.E.; Xia, Y. Inflammation, autoimmunity, and hypertension: The essential role of tissue transglutaminase. Am. J. Hypertens. 2017, 30, 756–764.
- Nicholas, B.; Smethurst, P.; Verderio, E.; Jones, R.; Griffin, M. Cross-linking of cellular proteins by tissue transglutaminase during necrotic cell death: A mechanism for maintaining tissue integrity. Biochem. J. 2003, 371, 413–422.
- Antonyak, M.A.; Miller, A.M.; Jansen, J.M.; Boehm, J.E.; Balkman, C.E.; Wakshlag, J.J.; Page, R.L.; Cerione, R.A. Augmentation of tissue transglutaminase expression and activation by epidermal growth factor inhibit doxorubicin-induced apoptosis in human breast cancer cells. J. Biol. Chem. 2004, 279, 41461–41467.
- Herman, J.F.; Mangala, L.S.; Mehta, K. Implications of increased tissue transglutaminase (TG2) expression in drug-resistant breast cancer (MCF-7) cells. Oncogene 2006, 25, 3049–3058.
- Park, K.S.; Kim, D.S.; Jeong, K.C.; Kim, S.Y. Increase in transglutaminase 2 expression is associated with NF-κB activation in breast cancer tissues. Front. Biosci. 2009, 14, 1945–1951.
- Ou, H.; Haendeler, J.; Aebly, M.R.; Kelly, L.A.; Cholewa, B.C.; Koike, G.; Kwitek-Black, A.; Jacob, H.J.; Berk, B.C.; Miano, J.M. Retinoic acid-induced tissue transglutaminase and apoptosis in vascular smooth muscle cells. Circ. Res. 2000, 87, 881–887.
- Shimada, J.; Suzuki, Y.; Kim, S.J.; Wang, P.C.; Matsumura, M.; Kojima, S. Transactivation via RAR/RXR-Sp1 interaction: Characterization of binding between Sp1 and GC box motif. Mol. Endocrinol. 2001, 15, 1677–1692.
- Ghanta, K.S.; Pakala, S.B.; Reddy, S.D.N.; Li, D.-Q.; Nair, S.S.; Kumar, R. MTA1 coregulation of transglutaminase 2 expression and function during inflammatory response. J. Biol. Chem. 2011, 286, 7132–7138.
- Ritter, S.J.; Davies, P.J.A. Identification of a transforming growth factor-β1/bone morphogenetic protein 4 (TGF-β1/BMP4) response element within the mouse tissue transglutaminase gene promoter. J. Biol. Chem. 1998, 273, 12798–12806.
- Mirza, A.; Liu, S.L.; Frizell, E.; Zhu, J.; Maddukuri, S.; Martinez, J.; Davies, P.; Schwarting, R.; Norton, P.; Zern, M.A. A role for tissue transglutaminase in hepatic injury and fibrogenesis, and its regulation by NF-kappaB. Am. J. Physiol. Liver Physiol. 1997, 272, G281–G288.
- Campisi, A.; Bramanti, V.; Caccamo, D.; Li Volti, G.; Cannavò, G.; Currò, M.; Raciti, G.; Galvano, F.; Amenta, F.; Vanella, A.; et al. Effect of growth factors and steroids on transglutaminase activity and expression in primary astroglial cell cultures. J. Neurosci. Res. 2008, 86, 1297–1305.
- Johnson, K.; Hashimoto, S.; Lotz, M.; Pritzker, K.; Terkeltaub, R. Interleukin-1 induces pro-mineralizing activity of cartilage tissue transglutaminase and factor XIIIa. Am. J. Pathol. 2001, 159, 149–163.
- Suto, N.; Ikura, K.; Sasaki, R. Expression induced by interleukin-6 of tissue-type transglutaminase in human hepatoblastoma HepG2 cells. J. Biol. Chem. 1993, 268, 7469–7473.
- Jang, G.Y.; Jeon, J.H.; Cho, S.Y.; Shin, D.M.; Kim, C.W.; Jeong, E.M.; Bae, H.C.; Kim, T.W.; Lee, S.H.; Choi, Y.; et al. Transglutaminase 2 suppresses apoptosis by modulating caspase 3 and NF-κB activity in hypoxic tumor cells. Oncogene 2010, 29, 356–367.
- Kuncio, G.S.; Tsyganskaya, M.; Zhu, J.; Liu, S.L.; Nagy, L.; Thomazy, V.; Davies, P.J.A.; Zern, M.A. TNF-α modulates expression of the tissue transglutaminase gene in liver cells. Am. J. Physiol. Gastrointest. Liver Physiol. 1998, 274.
- Jambrovics, K.; Uray, I.P.; Keresztessy, Z.; Keillor, J.W.; Fésüs, L.; Balajthy, Z. Transglutaminase 2 programs differentiating acute promyelocytic leukemia cells in all-trans retinoic acid treatment to inflammatory stage through NF-κB activation. Haematologica 2019, 104, 505–515.
- Balajthy, Z.; Csomós, K.; Vámosi, G.; Szántó, A.; Lanotte, M.; Fésüs, L. Tissue-transglutaminase contributes to neutrophil granulocyte differentiation and functions. Blood 2006, 108, 2045–2054.
- Singh, U.S.; Pan, J.; Kao, Y.L.; Joshi, S.; Young, K.L.; Baker, K.M. Tissue transglutaminase mediates activation of RhoA and MAP kinase pathways during retinoic acid-induced neuronal differentiation of SH-SY5Y cells. J. Biol. Chem. 2003, 278, 391–399.
- Tucholski, J.; Lesort, M.; Johnson, G.V.W. Tissue transglutaminase is essential for neurite outgrowth in human neuroblastoma SH-SY5Y cells. Neuroscience 2001, 102, 481–491.
- Liu, T.; Tee, A.E.L.; Porro, A.; Smith, S.A.; Dwarte, T.; Pei, Y.L.; Iraci, N.; Sekyere, E.; Haber, M.; Norris, M.D.; et al. Activation of tissue transglutaminase transcription by histone deacetylase inhibition as a therapeutic approach for Myc oncogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18682–18687.
- Carbone, C.; Di Gennaro, E.; Piro, G.; Milone, M.R.; Pucci, B.; Caraglia, M.; Budillon, A. Tissue transglutaminase (TG2) is involved in the resistance of cancer cells to the histone deacetylase (HDAC) inhibitor vorinostat. Amino Acids 2017, 49, 517–528.
- Quan, G.; Choi, J.Y.; Lee, D.S.; Lee, S.C. TGF-β1 up-regulates transglutaminase two and fibronectin in dermal fibroblasts: A possible mechanism for the stabilization of tissue inflammation. Arch. Dermatol. Res. 2005, 297, 84–90.
- Lisak, R.P.; Nedelkoska, L.; Studzinski, D.; Bealmear, B.; Xu, W.; Benjamins, J.A. Cytokines regulate neuronal gene expression: Differential effects of Th1, Th2 and monocyte/macrophage cytokines. J. Neuroimmunol. 2011, 238, 19–33.
- Frisdal, E.; Lesnik, P.; Olivier, M.; Robillard, P.; Chapman, M.J.; Huby, T.; Guerin, M.; Le Goff, W. Interleukin-6 protects human macrophages from cellular cholesterol accumulation and attenuates the proinflammatory response. J. Biol. Chem. 2011, 286, 30926–30936.
- Chen, Y.; Wang, Y.; Lin, W.; Sheng, R.; Wu, Y.; Xu, R.; Zhou, C.; Yuan, Q. AFF1 inhibits adipogenic differentiation via targeting TGM2 transcription. Cell Prolif. 2020, 53, e12831.
- Ribas, V.; Drew, B.G.; Le, J.A.; Soleymani, T.; Daraei, P.; Sitz, D.; Mohammad, L.; Henstridge, D.C.; Febbraio, M.A.; Hewitt, S.C.; et al. Myeloid-specific estrogen receptor α deficiency impairs metabolic homeostasis and accelerates atherosclerotic lesion development. Proc. Natl. Acad. Sci. USA 2011, 108, 16457–16462.
- Evans, M.J.; Harris, H.A.; Miller, C.P.; Karathanasis, S.K.; Adelman, S.J. Estrogen receptors α and β have similar activities in multiple endothelial cell pathways. Endocrinology 2002, 143, 3785–3795.
- Gundemir, S.; Colak, G.; Tucholski, J.; Johnson, G.V.W. Transglutaminase 2: A molecular Swiss army knife. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 406–419.
- Lu, S.; Saydak, M.; Gentile, V.; Stein, J.P.; Davies, P.J.A. Isolation and characterization of the human tissue transglutaminase gene promoter. J. Biol. Chem. 1995, 270, 9748–9756.
- Cellura, D.; Pickard, K.; Quaratino, S.; Parker, H.; Strefford, J.C.; Thomas, G.J.; Mitter, R.; Mirnezami, A.H.; Peake, N.J. miR-19-mediated inhibition of transglutaminase-2 leads to enhanced invasion and metastasis in colorectal cancer. Mol. Cancer Res. 2015, 13, 1095–1105.
- Sándor, K.; Daniel, B.; Kiss, B.; Kovács, F.; Szondy, Z. Transcriptional control of transglutaminase 2 expression in mouse apoptotic thymocytes. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 964–974.
- Bianchi, N.; Beninati, S.; Bergamini, C.M. Spotlight on the transglutaminase 2 gene: A focus on genomic and transcriptional aspects. Biochem. J. 2018, 475, 1643–1667.
- Lai, T.S.; Greenberg, C.S. TGM2 and implications for human disease: Role of alternative splicing. Front. Biosci. 2013, 18, 504–519.
- Mariani, P.; Carsughi, F.; Spinozzi, F.; Romanzetti, S.; Meier, G.; Casadio, R.; Bergamini, C.M. Ligand-induced conformational changes in tissue transglutaminase: Monte Carlo analysis of small-angle scattering data. Biophys. J. 2000, 78, 3240–3251.
- Stamnaes, J.; Pinkas, D.M.; Fleckenstein, B.; Khosla, C.; Sollid, L.M. Redox regulation of transglutaminase 2 activity. J. Biol. Chem. 2010, 285, 25402–25409.
- Jin, X.; Stamnaes, J.; Klöck, C.; DiRaimondo, T.R.; Sollid, L.M.; Khosla, C. Activation of extracellular transglutaminase 2 by thioredoxin. J. Biol. Chem. 2011, 286, 37866–37873.
- Lai, T.S.; Hausladen, A.; Slaughter, T.F.; Eu, J.P.; Stamler, J.S.; Greenberg, C.S. Calcium regulates S-nitrosylation, denitrosylation, and activity of tissue transglutaminase. Biochemistry 2001, 40, 4904–4910.
- Santhanam, L.; Tuday, E.C.; Webb, A.K.; Dowzicky, P.; Kim, J.H.; Oh, Y.J.; Sikka, G.; Kuo, M.; Halushka, M.K.; MacGregor, A.M.; et al. Decreased S-nitrosylation of tissue transglutaminase contributes to age-related increases in vascular stiffness. Circ. Res. 2010, 107, 117–125.
- Lai, T.S.; Davies, C.; Greenberg, C.S. Human tissue transglutaminase is inhibited by pharmacologic and chemical acetylation. Protein Sci. 2010, 19, 229–235.
- Luciani, A.; Villella, V.R.; Vasaturo, A.; Giardino, I.; Raia, V.; Pettoello-Mantovani, M.; D’Apolito, M.; Guido, S.; Leal, T.; Quaratino, S.; et al. SUMOylation of tissue transglutaminase as link between oxidative stress and inflammation. J. Immunol. 2009, 183, 2775–2784.
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Pettoello-Mantovani, M.; et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 2010, 12, 863–875.
- Grenard, P.; Bresson-Hadni, S.; El Alaoui, S.; Chevallier, M.; Vuitton, D.A.; Ricard-Blum, S. Transglutaminase-mediated cross-linking is involved in the stabilization of extracellular matrix in human liver fibrosis. J. Hepatol. 2001, 35, 367–375.
- Elli, L.; Bergamini, C.M.; Bardella, M.T.; Schuppan, D. Transglutaminases in inflammation and fibrosis of the gastrointestinal tract and the liver. Dig. Liver Dis. 2009, 41, 541–550.
- Tatsukawa, H.; Tani, Y.; Otsu, R.; Nakagawa, H.; Hitomi, K. Global identification and analysis of isozyme-specific possible substrates crosslinked by transglutaminases using substrate peptides in mouse liver fibrosis. Sci. Rep. 2017, 7, 45049.
- Wu, J.; Zern, M.A. Tissue transglutaminase, a key enzyme involved in liver diseases. Hepatol. Res. 2004, 29, 1–8.
- Verderio, E.A.M.; Furini, G.; Burhan, I.W.; Johnson, T.S. Transglutaminases: Expression in kidney and relation to kidney fibrosis. In Transglutaminases; Springer: Tokyo, Japan, 2015; pp. 229–262. ISBN 9784431558255.
- Johnson, T.S.; Griffin, M.; Thomas, G.L.; Skill, J.; Cox, A.; Yang, B.; Nicholas, B.; Birckbichler, P.J.; Muchaneta-Kubara, C.; El Nahas, A.M. The role of transglutaminase in the rat subtotal nephrectomy model of renal fibrosis. J. Clin. Investig. 1997, 99, 2950–2960.
- Johnson, T.S. Tissue transglutaminase and the progression of human renal scarring. J. Am. Soc. Nephrol. 2003, 14, 2052–2062.
- El Nahas, A.M.; Abo-Zenah, H.; Skill, N.J.; Bex, S.; Wild, G.; Griffin, M.; Johnson, T.S. Elevated ε-(γ-glutamyl)lysine in human diabetic nephropathy results from increased expression and cellular release of tissue transglutaminase. Nephron Clin. Pract. 2004, 97, c108–c117.
- Tatsukawa, H.; Otsu, R.; Tani, Y.; Wakita, R.; Hitomi, K. Isozyme-specific comprehensive characterization of transglutaminase-crosslinked substrates in kidney fibrosis. Sci. Rep. 2018, 8, 7306.
- Prat-Duran, J.; Pinilla, E.; Nørregaard, R.; Simonsen, U.; Buus, N.H. Transglutaminase 2 as a novel target in chronic kidney disease—Methods, mechanisms and pharmacological inhibition. Pharmacol. Ther. 2021, 222, 107787.
- Griffin, M.; Smith, L.L.; Wynne, J. Changes in transglutaminase activity in an experimental model of pulmonary fibrosis induced by paraquat. Br. J. Exp. Pathol. 1979, 60, 653–661.
- Olsen, K.C.; Sapinoro, R.E.; Kottmann, R.M.; Kulkarni, A.A.; Iismaa, S.E.; Johnson, G.V.W.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J. Transglutaminase 2 and its role in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 699–707.
- Takeuchi, T.; Tatsukawa, H.; Shinoda, Y.; Kuwata, K.; Nishiga, M.; Takahashi, H.; Hase, N.H.K. Spatially resolved identification of transglutaminase substrates 2 by proteomics in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, in press.
- Philp, C.J.; Siebeke, I.; Clements, D.; Miller, S.; Habgood, A.; John, A.E.; Navaratnam, V.; Hubbard, R.B.; Jenkins, G.; Johnson, S.R. Extracellular matrix cross-linking enhances fibroblast growth and protects against matrix proteolysis in lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 594–603.
- Collighan, R.J.; Griffin, M. Transglutaminase 2 cross-linking of matrix proteins: Biological significance and medical applications. Amino Acids 2009, 36, 659–670.
- Oh, K.; Park, H.B.; Byoun, O.J.; Shin, D.M.; Jeong, E.M.; Kim, Y.W.; Kim, Y.S.; Melino, G.; Kim, I.G.; Lee, D.S. Epithelial transglutaminase 2 is needed for T cell interleukin-17 production and subsequent pulmonary inflammation and fibrosis in bleomycin-treated mice. J. Exp. Med. 2011, 208, 1707–1719.
- Wang, Z.; Stuckey, D.J.; Murdoch, C.E.; Camelliti, P.; Lip, G.Y.H.; Griffin, M. Cardiac fibrosis can be attenuated by blocking the activity of transglutaminase 2 using a selective small-molecule inhibitor. Cell Death Dis. 2018, 9.
- Shinde, A.V.; Frangogiannis, N.G. Tissue transglutaminase in the pathogenesis of heart failure. Cell Death Differ. 2018, 25, 453–456.
- Kagan, H.M. Intra- and extracellular enzymes of collagen biosynthesis as biological and chemical targets in the control of fibrosis. Acta Trop. 2000, 77, 147–152.
- Avery, N.C.; Bailey, A.J. Restraining cross-links responsible for the mechanical properties of collagen fibers: Natural and artificial. In Collagen: Structure and Mechanics; Springer: Berlin/Heidelberg, Germany, 2008; pp. 81–110. ISBN 9780387739052.
- Simon, D.D.; Niklason, L.E.; Humphrey, J.D. Tissue transglutaminase, not lysyl oxidase, dominates early calcium-dependent remodeling of fibroblast-populated collagen lattices. Cells Tissues Organs 2014, 200, 104–117.
- Kojima, S.; Nara, K.; Rifkin, D.B. Requirement for transglutaminase in the activation of latent transforming growth factor-β in bovine endothelial cells. J. Cell Biol. 1993, 121, 439–448.
- Shweke, N.; Boulos, N.; Jouanneau, C.; Vandermeersch, S.; Melino, G.; Dussaule, J.-C.; Chatziantoniou, C.; Ronco, P.; Boffa, J.-J. Tissue transglutaminase contributes to interstitial renal fibrosis by favoring accumulation of fibrillar collagen through TGF-β activation and cell infiltration. Am. J. Pathol. 2008, 173, 631–642.
- Verderio, E.; Gaudry, C.; Gross, S.; Smith, C.; Downes, S.; Griffin, M. Regulation of cell surface tissue transglutaminase: Effects on matrix storage of latent transforming growth factor-β binding protein-1. J. Histochem. Cytochem. 1999, 47, 1417–1432.
- Telci, D.; Collighan, R.J.; Basaga, H.; Griffin, M. Increased TG2 expression can result in induction of transforming growth factor β1, causing increased synthesis and deposition of matrix proteins, which can be regulated by nitric oxide. J. Biol. Chem. 2009, 284, 29547–29558.
- Ichinose, A.; Bottenus, R.E.; Davie, E.W. Structure of transglutaminases. J. Biol. Chem. 1990, 265, 13411–13414.
- Akimov, S.S.; Krylov, D.; Fleischman, L.F.; Belkin, A.M. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J. Cell Biol. 2000, 148, 825–838.
- Verderio, E.A.M.; Telci, D.; Okoye, A.; Melino, G.; Griffin, M. A novel RGD-independent cell adhesion pathway mediated by fibronectin-bound tissue transglutaminase rescues cells from anoikis. J. Biol. Chem. 2003, 278, 42604–42614.
- Wang, Z.; Collighan, R.J.; Gross, S.R.; Danen, E.H.J.; Orend, G.; Telci, D.; Griffin, M. RGD-independent cell adhesion via a tissue transglutaminase-fibronectin matrix promotes fibronectin fibril deposition and requires syndecan-4/2 and α5β1 integrin co-signaling. J. Biol. Chem. 2010, 285, 40212–40229.
- Telci, D.; Wang, Z.; Li, X.; Verderio, E.A.M.; Humphries, M.J.; Baccarini, M.; Basaga, H.; Griffin, M. Fibronectin-tissue transglutaminase matrix rescues RGD-impaired cell adhesion through syndecan-4 and β1 integrin co-signaling. J. Biol. Chem. 2008, 283, 20937–20947.
- Scarpellini, A.; Huang, L.; Burhan, I.; Schroeder, N.; Funck, M.; Johnson, T.S.; Verderio, E.A.M. Syndecan-4 knockout leads to reduced extracellular transglutaminase-2 and protects against tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2014, 25, 1013–1027.
- Tatsukawa, H.; Fukaya, Y.; Frampton, G.; Martinez–Fuentes, A.; Suzuki, K.; Kuo, T.-F.; Nagatsuma, K.; Shimokado, K.; Okuno, M.; Wu, J.; et al. Role of transglutaminase 2 in liver injury via cross-linking and silencing of transcription factor Sp1. Gastroenterology 2009, 136, 1783–1795.e10.
- Tatsukawa, H.; Furutani, Y.; Hitomi, K.; Kojima, S. Transglutaminase 2 has opposing roles in the regulation of cellular functions as well as cell growth and death. Cell Death Dis. 2016, 7, e2244.
- Borowiak, M.; Garratt, A.N.; Wüstefeld, T.; Strehle, M.; Trautwein, C.; Birchmeier, C. Met provides essential signals for liver regeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 10608–10613.
- Huh, C.G.; Factor, V.M.; Sánchez, A.; Uchida, K.; Conner, E.A.; Thorgeirsson, S.S. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc. Natl. Acad. Sci. USA 2004, 101, 4477–4482.
- Kojima, S.; Kuo, T.-F.; Tatsukawa, H.; Hirose, S. Induction of cross-linking and silencing of Sp1 by transglutaminase during liver injury in ASH and NASH via different ER stress pathways. Dig. Dis. 2010, 28, 715–721.
- Popov, Y.; Sverdlov, D.Y.; Sharma, A.K.; Bhaskar, K.R.; Li, S.; Freitag, T.L.; Lee, J.; Dieterich, W.; Melino, G.; Schuppan, D. Tissue transglutaminase does not affect fibrotic matrix stability or regression of liver fibrosis in mice. Gastroenterology 2011, 140, 1642–1652.
- D’Argenio, G.; Amoruso, D.C.; Mazzone, G.; Vitaglione, P.; Romano, A.; Ribecco, M.T.; D’Armiento, M.R.; Mezza, E.; Morisco, F.; Fogliano, V.; et al. Garlic extract prevents CCl4-induced liver fibrosis in rats: The role of tissue transglutaminase. Dig. Liver Dis. 2010, 42, 571–577.
- Badarau, E.; Wang, Z.; Rathbone, D.L.; Costanzi, A.; Thibault, T.; Murdoch, C.E.; El Alaoui, S.; Bartkeviciute, M.; Griffin, M. Development of potent and selective tissue transglutaminase inhibitors: Their effect on TG2 function and application in pathological conditions. Chem. Biol. 2015, 22, 1347–1361.
- Nyabam, S.; Wang, Z.; Thibault, T.; Oluseyi, A.; Basar, R.; Marshall, L.; Griffin, M. A novel regulatory role for tissue transglutaminase in epithelial-mesenchymal transition in cystic fibrosis. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2234–2244.
- Olsen, K.C.; Epa, A.P.; Kulkarni, A.A.; Kottmann, R.M.; McCarthy, C.E.; Johnson, G.V.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J. Inhibition of transglutaminase 2, a novel target for pulmonary fibrosis, by two small electrophilic molecules. Am. J. Respir. Cell Mol. Biol. 2014, 50, 737–747.
- Johnson, T.S.; Fisher, M.; Haylor, J.L.; Hau, Z.; Skill, N.J.; Jones, R.; Saint, R.; Coutts, I.; Vickers, M.E.; El Nahas, A.M.; et al. Transglutaminase inhibition reduces fibrosis and preserves function in experimental chronic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 3078–3088.
- Qiu, J.F.; Zhang, Z.Q.; Chen, W.; Wu, Z.Y. Cystamine ameliorates liver fibrosis induced by carbon tetrachloride via inhibition of tissue transglutaminase. World J. Gastroenterol. 2007, 13, 4328–4332.
- Fell, S.; Wang, Z.; Blanchard, A.; Nanthakumar, C.; Griffin, M. Transglutaminase 2: A novel therapeutic target for idiopathic pulmonary fibrosis using selective small molecule inhibitors. Amino Acids 2021, 53, 205–217.


